Estructura de las proteínas
La estructura de las proteínas reúne las propiedades de disposición en el espacio de las moléculas de proteína que provienen de su secuencia de aminoácidos, las características físicas de su entorno y la presencia de compuestos simples o complejos que las estabilicen y conduzcan a un plegamiento específico.

Aminoácidos
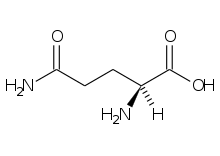
Los aminoácidos, monómeros componentes del polímero proteína, son moléculas quirales constituidas por un átomo de carbono central, el Cα, que portan en este un grupo amino y un grupo carboxilo, lo cual les da su nombre, además de un átomo de hidrógeno y una cadena lateral que les confiere sus características definitorias, y en función de la cual se clasifican.[1]
Los 20 α-L-aminoácidos proteinogénicos son los listados en la siguiente tabla:
| Nombre | Código de tres letras |
Código de una letra |
Abundancia relativa (%) E.C. |
MW | pK | VdW volumen (Å3) |
Cargado, Polar, Hidrofóbico, Neutro |
|---|---|---|---|---|---|---|---|
| Alanina | Ala | A | 13.0 | 71 | 67 | H | |
| Arginina | Arg | R | 5.3 | 157 | 12.5 | 148 | C+ |
| Asparagina | Asn | N | 9.9 | 114 | 96 | P | |
| Aspartato | Asp | D | 9.9 | 114 | 3.9 | 91 | C- |
| Cisteína | Cys | C | 1.8 | 103 | 86 | P | |
| Glutamato | Glu | E | 10.8 | 128 | 4.3 | 109 | C- |
| Glutamina | Gln | Q | 10.8 | 128 | 114 | P | |
| Glicina | Gly | G | 7.8 | 57 | 48 | N | |
| Histidina | His | H | 0.7 | 137 | 6.0 | 118 | P, C+ |
| Isoleucina | Ile | I | 4.4 | 113 | 124 | H | |
| Leucina | Leu | L | 7.8 | 113 | 124 | H | |
| Lisina | Lys | K | 7.0 | 129 | 10.5 | 135 | C+ |
| Metionina | Met | M | 3.8 | 131 | 124 | H | |
| Fenilalanina | Phe | F | 3.3 | 147 | 135 | H | |
| Prolina | Pro | P | 4.6 | 97 | 90 | H | |
| Serina | Ser | S | 6.0 | 87 | 73 | P | |
| Treonina | Thr | T | 4.6 | 101 | 93 | P | |
| Triptófano | Trp | W | 1.0 | 186 | 163 | P | |
| Tirosina | Tyr | Y | 2.2 | 163 | 10.1 | 141 | P |
| Valina | Val | V | 6.0 | 99 | 105 | H |
Termodinámica del plegamiento
En condiciones fisiológicas, el proceso de plegamiento de una proteína globular está claramente favorecido termodinámicamente, esto es, el incremento de energía libre global del proceso debe ser negativo (aunque alguno de sus pasos puede ser positivo). Este cambio se consigue equilibrando una serie de factores termodinámicos como la entropía conformacional, las interacciones carga-carga, los puentes de hidrógeno internos, las interacciones hidrofóbicas y las interacciones de van der Waals.[2] .[3]
Entropía conformacional
Se define entropía conformacional del plegado como la disminución de la entropía, de la aleatoriedad en definitiva, durante el paso desde una multitud de conformaciones de ovillo aleatorio hasta una única estructura plegada. La energía libre, representada en la ecuación ΔG = ΔH - T ΔS, demuestra que el ΔS negativo realiza una contribución positiva a ΔG. Es decir, el cambio de entropía conformacional se opone al plegado. Por ello, el ΔG global, que debe ser negativo, se debe a que o bien ΔH es negativo y grande o a algún otro aumento de la entropía con el plegado. En la práctica se dan ambas cosas.[3]
La fuente de ΔH negativo es el cúmulo de interacciones favorables energéticamente que se dan en el interior del glóbulo proteico, interacciones que suelen ser no covalentes.
Interacciones carga-carga
Las interacciones carga-carga se dan entre grupos polares y cargados de las cadenas laterales de los aminoácidos componentes del polipéptido, puesto que los grupos carboxilo y amino del carbono alfa están implicados en el enlace peptídico. De este modo, dichos grupos ionizados se atraen y forman un equivalente a sales entre residuos del polipéptido: de hecho, se denominan a veces puentes salinos.[3] Evidentemente, dichas interacciones desaparecen cuando el pH del medio es tal que se pierde el estado de ionización del grupo; de hecho, esta es una de las causas de la desnaturalización rápida de las proteínas mediante adición de ácidos o bases, y subyuga a las proteínas a un entorno de un pH tamponado y moderado, que es el fisiológico, salvo excepciones, como puede ser el interior lisosomal en el entorno subcelular[4]
Enlaces de hidrógeno internos

Las cadenas laterales de muchos aminoácidos se comportan como donadores o como aceptores de enlaces de hidrógeno (es el caso de los grupos hidroxilo de la serina y los grupos amino de la glutamina, por ejemplo). Además, si los protones amida o los carbonilos del armazón polipeptídico no están implicados en el enlace peptídico, pueden interaccionar también en este tipo de uniones estabilizadoras.
Si bien los enlaces de hidrógeno son débiles en disolución acuosa.[3] su gran número puede estabilizar, y lo hace, la estructura terciaria proteica.
Interacciones de van der Waals
El denso empaquetamiento en el núcleo de las proteínas globulares facilita la interacción débil entre grupos moleculares sin carga. Dichos enlaces son de baja energía, pero su abundante número suple su debilidad. Cada interacción individual sólo contribuye en unos pocos kilojulios a la entalpía de interacción negativa global. Pero la suma de todas las contribuciones de todas las interacciones sí que puede estabilizar a la estructura plegada. De este modo, una contribución energética favorable a partir de la suma de las interacciones intramoleculares compensa de modo más que suficiente la entropía desfavorable del plegado.[3]
Interacciones hidrofóbicas
Por definición, cualquier sustancia hidrofóbica en contacto con el agua provoca que ésta huya y se agrupe en estructuras denominadas clatratos.[4] Esta ordenación corresponde a una disminución de la entropía del sistema. En el caso de las proteínas, los residuos hidrofóbicos de los aminoácidos quedan orientados, en su plegamiento, hacia el interior de la molécula, en contacto con sus semejantes y alejados del agua. En consecuencia, la internalización de los grupos hidrófobos aumenta la aleatoriedad del sistema 'proteína más agua' y, por consiguiente, produce un aumento de entropía al doblarse. Este aumento de entropía produce una contribución negativa a la energía libre del plegado y aumenta la estabilidad de la estructura proteica.[3]
Función de los enlaces disulfuro
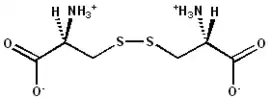
La estabilización de la proteína recién plegada puede suceder mediante la formación de puentes disulfuro entre residuos de cisteína adyacentes o enfrentados. Dicho procesamiento se produce en muchos casos en el lumen del retículo endoplasmático mediante la enzima disulfuro isomerasa, presente en todas las células eucariotas. Dicha enzima, que cataliza la oxidación de los grupos sulfhidrilo o grupos tiol (-SH2) de los residuos de cisteína, es especialmente abundante en órganos como el hígado o páncreas, donde se producen pequeñas cantidades de proteínas que contienen este tipo de enlaces.[5]
Niveles de estructuración

La estructura de las proteínas puede jerarquizarse en una serie de niveles, interdependientes. Estos niveles corresponden a:
- Estructura primaria, que corresponde a la secuencia de aminoácidos unidos en fila.
- Estructura secundaria, que provoca la aparición de motivos estructurales.
- Estructura terciaria, que define la estructura de las proteínas compuestas por un solo polipéptido.
- Estructura cuaternaria, si interviene más de un polipéptido.
Estructura primaria
La estructura primaria de las proteínas se refiere a la secuencia de aminoácidos, es decir, la combinación lineal de los aminoácidos mediante un tipo de enlace covalente, el enlace peptídico. Los aminoácidos están unidos por enlaces peptídicos siendo una de sus características más importantes la coplanaridad de los radicales constituyentes del enlace.
La estructura lineal del péptido definirá en gran medida las propiedades de niveles de organización superiores de la proteína. Este orden es consecuencia de la información del material genético: Cuando se produce la traducción del RNA se obtiene el orden de aminoácidos que van a dar lugar a la proteína. Se puede decir, por tanto, que la estructura primaria de las proteínas no es más que el orden de aminoácidos que la conforman.
Estructura secundaria
La estructura secundaria de las proteínas es la disposición espacial local del esqueleto proteico, gracias a la formación de puentes de hidrógeno entre los átomos que forman el enlace peptídico, es decir, un tipo de enlace covalente, sin hacer referencia a la cadena lateral. Existen diferentes tipos de estructura secundaria: - Estructura secundaria ordenada, ( repetitivos donde se encuentran los hélices alfa y cadenas beta, y no repetitivos donde se encuentran los giros beta y comba beta) -Estructura secundaria no ordenada -Estructura secundaria desordenada
Los motivos más comunes son la hélice alfa y la lámina beta (hoja plegada beta).
- Hélice alfa
Los aminoácidos en una hélice alfa están dispuestos en una estructura helicoidal dextrógira, con unos 3.6 aminoácidos por vuelta. Cada aminoácido supone un giro de unos 100° en la hélice, y los carbonos alfa de dos aminoácidos contiguos están separados por 1.5 Å. La hélice está estrechamente empaquetada, de forma que no hay casi espacio libre dentro de la hélice. Todas las cadenas laterales de los aminoácidos están dispuestas hacia el exterior de la hélice.[6]
El grupo amino del aminoácido (n) puede establecer un enlace de hidrógeno con el grupo carboxilo del aminoácido (n+4). De esta forma, cada aminoácido (n) de la hélice forma dos puentes de hidrógeno con su enlace peptídico y el enlace peptídico del aminoácido en (n+4) y en (n-4). En total son 7 enlaces de hidrógeno por vuelta. Esto estabiliza enormemente la hélice. Está dentro de los niveles de organización de la proteína.
- Lámina beta
La beta lámina se forma por el posicionamiento paralelo de dos cadenas de aminoácidos dentro de la misma proteína, en el que los grupos amino de una de las cadenas forman enlaces de hidrógeno con los grupos carboxilo de la opuesta. Es una estructura muy estable que puede llegar a resultar de una ruptura de los enlaces de hidrógeno durante la formación de la hélice alfa. Las cadenas laterales de esta estructura están posicionados sobre y bajo el plano de las láminas. Dichos sustituyentes no deben ser muy grandes, ni crear un impedimento estérico, ya que se vería afectada la estructura de la lámina.[7]
Estructura terciaria
Es el modo en que la cadena polipeptídica se pliega en el espacio, es decir, cómo se enrolla una determinada proteína, ya sea globular o fibrosa. Es la disposición de los dominios en el espacio.
La estructura terciaria se realiza de manera que los aminoácidos apolares se sitúan hacia el interior y los polares hacia el exterior en medios acuosos. Esto provoca una estabilización por interacciones hidrofóbicas, de fuerzas de van der Waals y de puentes disulfuro[1] (covalentes, entre aminoácidos de cisteína convenientemente orientados) y mediante enlaces iónicos.
Estructura cuaternaria
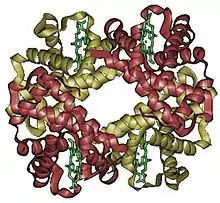
La estructura cuaternaria deriva de la conjunción de varias cadenas peptídicas que, asociadas, conforman un ente, un multímero, que posee propiedades distintas a la de sus monómeros componentes. Dichas subunidades se asocian entre sí mediante interacciones no covalentes, como pueden ser puentes de hidrógeno, interacciones hidrofóbicas o puentes salinos. Para el caso de una proteína constituida por dos monómeros, un dímero, este puede ser un homodímero, si los monómeros constituyentes son iguales, o un heterodímero, si no lo son.
Ángulos de rotación y representaciones de Ramachandran
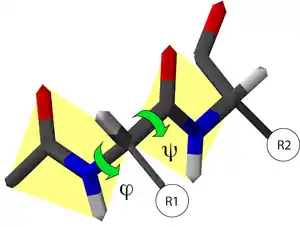
En una cadena polipeptídica, se definen dos enlaces del armazón capaces de rotar: uno es el enlace entre el nitrógeno y el Cα, y el otro el enlace entre el Cα y el carbono del grupo carbonilo. Ambos definen dos ángulos de rotación:
- El ángulo de rotación φ, definido por los átomos CO-NH-Cα-CO; el primer y el último carbono de la secuencia pertenecen a dos aminoácidos consecutivos.
- El ángulo de rotación ψ, definido por los átomos NH-Cα-CO-NH; los dos nitrógenos en esta secuencia pertenecen a dos aminoácidos consecutivos.
La orientación del eje de giro considerada positiva por convención se muestra en la imagen; corresponde a la propia de las agujas del reloj.[3]
Existen dos excepciones en los aminoácidos que se representan en estos diagramas: la glicina, carente de un sustituyente, y la prolina, cíclica debido a la tenencia de una estructura tipo pirrol, no cumplen los requisitos requeridos para una representación convencional[8]

Se puede describir, por tanto, la conformación del armazón de cualquier residuo concreto de una proteína especificando estos dos ángulos.[3] Con estos dos parámetros podemos describir la conformación de dicho residuo en un mapa mediante un punto, con coordenadas φ y ψ. Para determinados tipos de estructura secundaria, como la hélice alfa, todos los residuos comparten dichos ángulos, por lo que un punto en el mapa en determinada posición puede describir una estructura secundaria. Estos mapas, denominados representaciones de Ramachandran, por el bioquímico G. N. Ramachandran[9] que los usó por ampliamente en [1963], permiten deducir dichas conformaciones.[3]
Dominios y Motivos
Es común que algunas zonas de la proteína tengan entidad estructural independiente, y a menudo funciones bioquímicas específicas, como, por ejemplo, alguna actividad catalítica. Su naturaleza depende de las estructuras anteriormente citadas a todos los niveles.
La estructura cuaternaria deriva de la conjunción de varias cadenas peptídicas que, asociadas, conforman un ente, un multímero, con propiedades distintas.
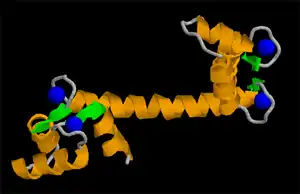
Las proteínas están organizadas en muchas unidades. Un dominio estructural es un elemento de la estructura de las proteínas que se autoestabiliza y a menudo estabiliza a los motivos conformacionales independientemente del resto de la cadena de proteína. Muchos dominios son únicos y proceden de una secuencia única de un gen o una familia génica pero en cambio otros aparecen en una variedad de proteínas. Los dominios son, a menudo, seleccionados evolutivamente porque poseen una función prominente en la biología de la proteína pertenecen; por ejemplo, "el domino de unión a calcio de calmodulina". La ingeniería genética permite modificar los dominios de una proteína a otra para generar proteínas quiméricas con funciones novedosas.[10] Un motivo en este sentido se refiere a una combinación específica de elementos estructurales secundarios (como los hélice-giro-hélice). Estos elementos son llamados a menudo superestructuras secundarias.
Suele denominarse motivo conformacional de forma global a un tipo de motivo, como los barriles-beta. La estructura de los motivos a menudo consiste en solo unos pocos elementos, por ejemplo, las hélice-giro-hélice, que sólo tienen tres. Se denota que la “secuencia espacial” es la misma en todas las instancias del motivo. Su orden es bastante irregular dentro del gen subyacente. Los motivos estructurales de la proteína a menudo incluyen giros de longitud variable en estructuras indeterminadas, lo que en efecto crea la plasticidad necesaria para unir dos elementos en el espacio que no están codificados por una secuencia de ADN inmediatamente adyacente en un gen. Se denota también que incluso cuando están codificados los elementos estructurales secundarios de un motivo en el mismo orden en dos genes, la composición cuantitativa de aminoácidos puede variar. Esto no sólo es cierto debido a las complicadas relaciones entre la estructura terciaria y primaria, sino por cuestiones relativas al tamaño. Si bien en la base de datos de levadura hay descritas unas 6.000 proteínas,[11] hay muchos menos dominios, motives estructurales y pliegues. Esto es, en parte, consecuencia de la evolución. Esto significa, por ejemplo, que un dominio de una proteína puede ser trasladado de una a otra, dando así una nueva función a las proteínas. Debido a estos mecanismos, los dominios o motivos estructurales pueden ser comunes a varias familias de proteínas.
Cinética del plegado de las proteínas
Aunque el plegamiento de las proteínas parta de un estado lineal inicial y uno final bien definidos, el proceso de plegamiento no es algo brusco con un lugar de partida y un fin, sino que está plagado de intermedios temporalmente mensurables y de vital importancia. Incluso, la estructura final dista mucho de ser estática: algunos autores imaginan a las proteínas como entidades dinámicas que continuamente cambian de estructura, de un modo similar al latido cardíaco[12]
Si bien el plegamiento de una proteína es un suceso rápido, que se completa en apenas un segundo, topológicamente es un problema muy complejo. Este hecho dio lugar a la paradoja de Levinthal, propia de Cyrus Levinthal en 1968: un cálculo aproximado indica que una cadena polipeptídica de unos 120 residuos posee unas 1050 conformaciones. Aunque la molécula pudiera intentar una nueva conformación cada 10-13 segundos, se necesitarían unos 1030 años para intentar un número significativo de ellas.[3] No obstante, proteínas de estas características se pliegan in vitro en un minuto.
La resolución de la paradoja pasa por la aceptación de la existencia de estados de plegamiento intermedios, en los que la proteína se encuentra parcialmente desplegada, en una ruta como sigue:[3]
- Proteína desplegada.
- Nucleación del plegado.
- Estados intermedios.
- Estado de glóbulo fundido.
- Reordenamientos finales.
- Proteína plegada.
Elementos moduladores
Temperatura
El papel de la temperatura es crucial puesto que su entidad físico-química, la energía cinética contenida en los átomos, dota de reactividad a los aminoácidos y, por ello, a las proteínas. No obstante, existe un límite, a unos 50 °C, sobrepasado el cual las proteínas pierden su conformación, esto es, se desnaturalizan.
La desnaturalización, producida por la temperatura y otros agentes desnaturalizantes, ocurre a varios niveles:
- En la desnaturalización de la estructura cuaternaria, las subunidades de proteínas se separan o su posición espacial se corrompe.
- La desnaturalización de la estructura terciaria implica la interrupción de:
- Enlaces covalentes entre las cadenas laterales de los aminoácidos (como los puentes disulfuro entre las cisteínas).
- Enlaces no covalentes dipolo-dipolo entre cadenas laterales polares de aminoácidos.
- Enlaces dipolo inducidos por fuerzas de Van Der Waals entre cadenas laterales no polares de aminoácidos.
- En la desnaturalización de la estructura secundaria las proteínas pierden todos los patrones de repetición regulares como las alfa hélices y adoptan formas aleatorias.
- La estructura primaria, la secuencia de aminoácidos ligados por enlaces peptídicos, no es interrumpida por las desnaturalización.
pH
El pH afecta al estado iónico de los aminoácidos, zwitteriones en definitiva, que no tienen implicado su grupo amino ni carboxilo en el enlace peptídico y, especialmente, a aquellos polares, con algún grupo cargado en su cadena lateral. El estado de ionización de este afecta a la reactividad y posibilidad, por tanto, de producir un enlace químico.[1]
Chaperonas
Las proteínas tipo chaperona son un conjunto de proteínas presentes en todas las células cuya función es la de ayudar al plegamiento de otras proteínas, tras su síntesis o durante su ciclo de actividad (por ejemplo, en defensa de estrés térmico).[4] Estas chaperonas no forman parte de la estructura primaria de la proteína funcional, sino que sólo se unen a ella para ayudar en su plegamiento, ensamblaje y transporte celular a otra parte de la célula donde la proteína realiza su función. Los cambios de conformación tridimensional de las proteínas pueden estar afectados por un conjunto de varias chaperonas que trabajan coordinadas, dependiendo de su propia estructura y de la disponibilidad de las chaperonas.[13]
Existen sustancias químicas no proteicas que pueden estabilizar a las proteínas mediante el establecimiento de enlaces de puente de hidrógeno, en sustitución a los del agua. Por ejemplo, es el caso de la trehalosa, un azúcar que se emplea en biotecnología para favorecer la viabilidad de las soluciones ricas en proteína incluso en condiciones de estrés ambiental[14]
Las chaperonas, localizadas en todos los compartimentos celulares, se agrupan en dos familias generales.[4]
- Chaperonas moleculares
Que se unen y estabilizan a proteínas desplegadas o parcialmente plegadas, evitando así que se agrupen y que sean degradadas. Integran la familia de las Hsp70 en el citosol y la matriz de la mitocondria, BiP en el retículo endoplasmático y DnaK en las bacterias.
- Chaperoninas
Que facilitan directamente el plegado de las proteínas. Incluyen a: TriC, en eucariotas; y GroEL, en bacterias y cloroplastos,
Clasificación estructural
Muchos métodos han sido desarrollados para la clasificación estructural de las proteínas; la recopilación de datos es almacenada en el Banco de Datos de Proteínas. Muchas bases de datos existentes clasifican las proteínas usando diferentes métodos. El SCOP, CATH y FSSP son los más usados.
Los métodos usados podrían clasificarse en: puramente manuales, manuales y automáticos y puramente automáticos. El mayor problema de estos métodos es la integración de los datos. La clasificación es constante entre SCOP, CATH Y FSSP para la mayoría de las proteínas que han sido clasificadas, pero hay todavía algunas diferencias e inconsistencias.
Determinación de la estructura proteica
Alrededor del 90% de las estructuras de las proteínas disponibles en el Banco de Datos de Proteínas han sido determinadas por cristalografía de rayos X. Este método permite medir la densidad de distribución de los electrones de la proteína en las 3 dimensiones (en el estado de cristalización), lo que permite obtener las coordenadas 3D de todos los átomos para determinar su posición con certeza. Aproximadamente el 9% de las estructuras de proteínas conocidas han sido obtenidas por técnicas de resonancia magnética nuclear, que también pueden ser usadas para identificar estructuras secundarias.

Nótese que los aspectos de las estructuras secundarias pueden ser detectados mediante medios bioquímicos como el dicroísmo circular[15] El microscopio crioelectrónico se ha convertido recientemente en un medio para determinar las estructuras proteicas con una resolución baja (menos de 5 Å o 0,5 nm) y se prevé que será una herramienta importante para los trabajos de alta resolución en la década próxima. Esta técnica es aún un recurso importante para científicos que están trabajando en complejos muy grandes de proteínas, como la cubierta y cápside de los virus y las proteínas amiloideas.[16][17]
| Resolución | Interpretación |
| >4.0 | Coordenadas individuales sin significado |
| 3.0 - 4.0 | Plegamiento posiblemente correcto, pero comúnmente con errores. Algunas cadenas laterales poseen mal los rotámeros. |
| 2.5 - 3.0 | Plegamiento bien dilucidado salvo en algunos pliegues superficiales, mal modelados. Algunas cadenas laterales largas (Lys, Glu, Gln) y otras cortas (Ser, Val, Thr) mal orientadas. |
| 2.0 - 2.5 | El número de cadenas laterales con un rotámero incorrecto es mucho menor. Los errores, pequeños, son detectados normalmente. Los pliegues superficiales están bastante bien definidos. Los ligandos y el agua son visibles. |
| 1.5 - 2.0 | Pocos residuos poseen mal rotámero. Los errores pequeños son detectados. Los pliegues incorrectos son muy raros, incluso en superficie. |
| 0.5 - 1.5 | En general, todo está correctamente resuelto. Las librerías de rotámeros y los estudios geométricos se hacen a este nivel de precisión. |
Investigación
Existen multitud de grupos que investigan todo lo relacionado con la determinación de la estructura proteica, su termodinámica, cinética e implicaciones. Estas últimas son de especial relevancia en neuropatología, puesto que enfermedades degenerativas como el Alzheimer[18] o infecciosas como el Creutzfeldt-Jacob[19] tienen su causa en alteraciones en la estructura de las proteínas.
Las herramientas de investigación son, en muchos casos, simulaciones de plegamiento mediante programas informáticos. Los proyectos de mayor actividad en cuanto a computación distribuida son:
- Docking@home (Universidad de Texas)
- Folding@homeArchivado el 8 de septiembre de 2012 en Wayback Machine. (Universidad de Stanford)
- Proteins@home (Escuela Politécnica de Francia)
- Rosetta@home (Universidad de Washington)
AlphaFold de Google Deepmind actualmente en el primer lugar del ranking CASP.
DeepMind y EMBL (Laboratorio Europeo de Biología Molecular), publican la predicción más de 350 000 estructuras de proteínas 3D[20][21]
Véase también
Referencias
- Lehninger, Albert (1993). Principles of Biochemistry, 2nd Ed. Worth Publishers. ISBN 0-87901-711-2.
- Pickett, S.D.; Sternberg, M.J. (1993), «Empirical scale of side-chain conformational entropy in protein folding» (w), J Mol Biol 231 (3): 825-39, doi:10.1006/jmbi.1993.1329.
- Mathews, C. K.; Van Holde, K.E et Ahern, K.G (2003). «6». Bioquímica (3ª edición). pp. 204 y ss. ISBN 978-84-7892-053-2.
- Lodish et al. (2005). Biología celular y molecular. Buenos Aires: Médica Panamericana. ISBN 950-06-1974-3
|isbn=incorrecto (ayuda). - Sela M, Lifson S. (1959). «The reformation of disulfide bridges in proteins». Biochim Biophys Acta 36 (2): 471-8. PMID 14444674. doi:10.1016/0006-3002(59)90188-X.
- Neurath, H (1940). «Intramolecular folding of polypeptide chains in relation to protein structure». Journal of Physical Chemistry 44: 296-305. doi:10.1021/j150399a003.
- Voet, Donald; Voet, Judith G. (2004). Biochemistry (3rd edición). Hoboken, NJ: Wiley. pp. 227-231. ISBN 047119350X.
- Carl-Ivar Brändén, John Tooze (trad. Bernard Lubochinsky, préf. Joël Janin), Introduction à la structure des protéines, De Boeck Université, Bruxelles, 1996 ISBN 2-8041-2109-7
- Ramachandran, G.N., Sasisekharan, V. & Ramakrishnan, C. (1963) J. Mol. Biol. 7, 95–99 (1963
- Casado-Vela, J. (12 de diciembre de 2012). «Protein chimerism: Novel source of protein diversity in humans adds complexity to bottom-up proteomics». Proteomics 13 (1): 5-11. doi:10.1002/pmic.201200371.
- Payne, W.E.; Garrels, J.I. (1997), «Yeast Protein database (YPD): a database for the complete proteome of Saccharomyces cerevisiae», Nucleic Acids Research 25 (1): 57-62, PMID 9016505, doi:10.1093/nar/25.1.57.
- Alberts et al (2004). Biología molecular de la célula. Barcelona: Omega. ISBN 8428213518.
- Ellis RJ, van der Vies SM (1991). «Molecular chaperones». Annu. Rev. Biochem. 60: 321-47. PMID 1679318. doi:10.1146/annurev.bi.60.070191.001541.
- Prescott, L.M. (199). Microbiología. McGraw-Hill Interamericana de España, S.A.U. ISBN 84-486-0261-7.
- Juan Antonio Lugo-Ríos. «Dicroismo circular». Archivado desde el original el 27 de octubre de 2007. Consultado el 8 de octubre de 2007.
- Chen, Shaoxia; Roseman, Alan M.; Hunter, Allison S.; Wood, Stephen P.; Burston, Steven G.; Ranson, Neil A.; Clarke, Anthony R.; Saibil, Helen R. (1994), «Location of a folding protein and shape changes in GroEL�GroES complexes imaged by cryo-electron microscopy» (w), Nature 371 (6494): 261-264, doi:10.1038/371261a0.
- Adrian, Marc; Dubochet, Jacques; Lepault, Jean; McDowall, Alasdair W. (1984), «Cryo-electron microscopy of viruses», Nature 308 (5954): 32-36, doi:10.1038/308032a0.
- Hashimoto M, Rockenstein E, Crews L, Masliah E (2003). «Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases.». Neuromolecular Med 4 (1-2): 21-36. PMID 14528050.
- Baker & Ridley (1996). Prion Disease. New Jersey: Humana Press. 0-89603-342-2.
- «DeepMind and EMBL release the most complete database of predicted 3D structures of human proteins». EMBL (en inglés estadounidense). 22 de julio de 2021. Consultado el 23 de julio de 2021.
- Tunyasuvunakool, Kathryn; Adler, Jonas; Wu, Zachary; Green, Tim; Zielinski, Michal; Žídek, Augustin; Bridgland, Alex; Cowie, Andrew et al. (22 de julio de 2021). «Highly accurate protein structure prediction for the human proteome». Nature (en inglés): 1-9. ISSN 1476-4687. doi:10.1038/s41586-021-03828-1. Consultado el 23 de julio de 2021.
| Control de autoridades |
|
|---|
 Datos: Q735188
Datos: Q735188 Multimedia: Protein structures / Q735188
Multimedia: Protein structures / Q735188