Superantígeno
Los superantígenos (SAgs) son una clase de antígenos que provocan una activación excesiva del sistema inmunitario. Específicamente, provoca la activación no específica de las células T, lo que resulta en la activación policlonal de las células T y la liberación masiva de citocinas. Los SAgs son producidos por algunos virus y bacterias patógenos, muy probablemente como mecanismo de defensa contra el sistema inmune.[1] En comparación con una respuesta normal de células T inducida por antígeno donde el 0,0001-0,001% de las células T del cuerpo están activadas, estos SAgs son capaces de activar hasta el 20% de las células T del cuerpo.[2] Además, los anticuerpos anti- CD3 y anti- CD28 (CD28-SuperMAB) también han demostrado ser altamente potentes superantígenos (y pueden activar hasta el 100% de las células T).
La gran cantidad de células T activadas genera una respuesta inmune masiva que no es específica de ningún epítopo en particular en el SAg, lo que socava una de las fortalezas fundamentales del sistema inmune adaptativo, es decir, su capacidad para atacar antígenos con alta especificidad. Más importante aún, la gran cantidad de células T activadas secretan grandes cantidades de citocinas, la más importante de las cuales es el interferón gamma. Este exceso de IFN-gamma a su vez activa los macrófagos. Los macrófagos activados, a su vez, sobreproducen citocinas proinflamatorias como IL-1, IL-6 y TNF-alfa. TNF-alfa es particularmente importante como parte de la respuesta inflamatoria del cuerpo. En circunstancias normales, se libera localmente en niveles bajos y ayuda al sistema inmunitario a vencer a los patógenos. Sin embargo, cuando se libera sistémicamente en la sangre y en niveles altos (debido a la activación masiva de las células T como resultado de la unión de SAg), puede causar síntomas graves y potencialmente mortales, como shock e insuficiencia orgánica múltiple.
Estructura
Los SAgs son producidos intracelularmente por bacterias y son liberados luego de la infección, como toxinas extracelulares maduras.[3]
Las secuencias de estas toxinas están relativamente conservadas entre los diferentes subgrupos. Más importante que la homología de secuencia, la estructura 3D es muy similar entre diferentes SAgs, lo que resulta en efectos funcionales similares entre diferentes grupos.[4][5]
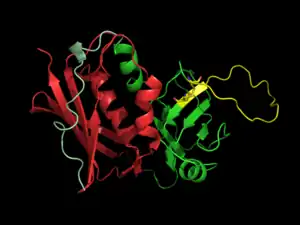
Las estructuras cristalinas de las enterotoxinas revelan que son proteínas elipsoidales compactas que comparten un patrón de plegamiento característico de dos dominios que comprende un dominio globular de barril β de terminal NH2 conocido como pliegue de oligosacárido/oligonucleótido, una hélice α larga que se extiende diagonalmente en el centro de la molécula, y un dominio globular terminal COOH.[4]
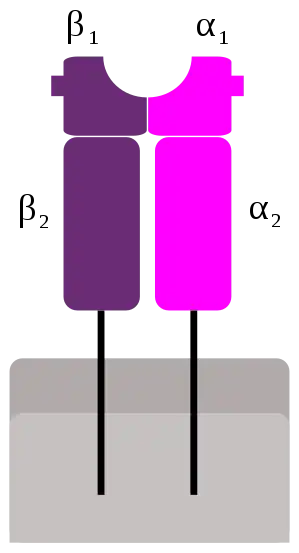
Los dominios tienen regiones de unión para el complejo principal de histocompatibilidad clase II (MHC clase II) y el receptor de células T (TCR), respectivamente.[6]
Unión
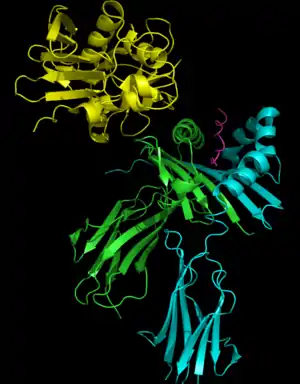
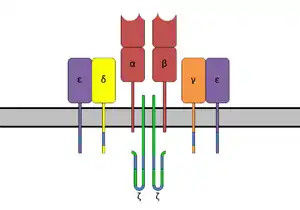
Los superantígenos se unen primero al MHC de clase II y luego se coordinan con la cadena alfa o beta variable de los receptores de células T (TCR).[5][7][8]
MHC Clase II
Los SAgs muestran preferencia por la forma HLA-DQ de la molécula.[8] La unión a la cadena α coloca el SAg en la posición adecuada para coordinar con el TCR.
Con menos frecuencia, los SAgs se unen a la cadena β polimórfica de MHC de clase II en una interacción mediada por un complejo de coordinación de iones de zinc entre tres residuos de SAg y una región altamente conservada de la cadena β de HLA-DR.[5] El uso de un ion de zinc en la unión conduce a una interacción de mayor afinidad.[4] Varios SAgs estafilocócicos son capaces de reticular moléculas de MHC uniéndose a las cadenas α y β. Este mecanismo estimula la expresión y liberación de citocinas en las células presentadoras de antígeno, así como también induce la producción de moléculas coestimuladoras que permiten que la célula se una y active las células T de manera más efectiva.
Receptor de células T
La región de unión a células T del SAg interactúa con la región variable en la cadena Beta del receptor de células T. Un SAg dado puede activar una gran proporción de la población de células T porque el repertorio de células T humanas comprende solo alrededor de 50 tipos de elementos Vβ y algunos SAgs son capaces de unirse a múltiples tipos de regiones Vβ. Esta interacción varía ligeramente entre los diferentes grupos de SAgs.[6] La variabilidad entre diferentes personas en los tipos de regiones de células T que prevalecen explica por qué algunas personas responden más fuertemente a ciertos SAG. El grupo I SAgs contacta con el Vβ en la región CDR2 y el marco de la molécula.[9][10] Los SAgs del Grupo II interactúan con la región Vβ utilizando mecanismos que dependen de la conformación. Estas interacciones son en su mayor parte independientes de cadenas laterales de aminoácidos Vβ específicos. Se ha demostrado que los SAg del grupo IV se dedican a los tres bucles CDR de ciertas formas Vβ. La interacción tiene lugar en una hendidura entre los dominios pequeño y grande del SAg y permite que el SAg actúe como una cuña entre el TCR y el MHC. Esto desplaza el péptido antigénico lejos del TCR y evita el mecanismo normal para la activación de las células T.[5][11]
La fuerza biológica del SAg (su capacidad de estimular) está determinada por su afinidad por el TCR. Los SAgs con la mayor afinidad por el TCR provocan la respuesta más fuerte.[12] El SPMEZ-2 es el SAg más potente descubierto hasta la fecha.
Señalización de células T
El SAg entrecruza el MHC y el TCR induciendo una ruta de señalización que resulta en la proliferación de la célula y la producción de citocinas. Esto ocurre porque un antígeno afín activa una célula T no por su estructura per se, sino porque su afinidad le permite unirse al TCR durante un período de tiempo lo suficientemente prolongado, y el SAg imita este enlace temporal. Se han encontrado niveles bajos de Zap-70 en las células T activadas por SAgs, lo que indica que la vía de señalización normal de la activación de las células T está deteriorada.[13]
Se presume que Fyn en lugar de Lck es activado por una tirosina quinasa, lo que conduce a la inducción adaptativa de la anergia.[14]
Tanto la ruta de la proteína quinasa C como la ruta de la proteína tirosina quinasa se activan, lo que da como resultado una producción reguladora de citocinas proinflamatorias.[15]
Esta vía de señalización alternativa deteriora ligeramente las vías de calcio/calcineurina y Ras/MAPquinasa,[14] pero permite una respuesta inflamatoria focalizada.
Efectos directos
La estimulación SAg de las células presentadoras de antígeno y las células T provoca una respuesta que es principalmente inflamatoria, centrada en la acción de las células Th1 T-helper. Algunos de los principales productos son IL-1, IL-2, IL-6, TNF-α, interferón gamma (IFN-γ), proteína inflamatoria de macrófagos 1α (MIP-1α), MIP-1β y proteína quimioatrayente de monocitos 1 (MCP-1).[15]
Esta liberación excesiva y descoordinada de citocinas (especialmente TNF-α) sobrecarga el cuerpo y produce erupciones cutáneas, fiebre y puede provocar insuficiencia multiorgánica, coma y muerte.[8][10]
La deleción o anergia de las células T activadas sigue a la infección. Esto resulta de la producción de IL-4 e IL-10 de la exposición prolongada a la toxina. La IL-4 y la IL-10 regulan negativamente la producción de IFN-gamma, MHC Clase II y moléculas coestimuladoras en la superficie de las APC. Estos efectos producen células de memoria que no responden a la estimulación antigénica.[16][17]
Un mecanismo por el cual esto es posible implica la supresión de las células T mediada por citoquinas. La reticulación de MHC también activa una vía de señalización que suprime la hematopoyesis y aumenta la apoptosis mediada por Fas.[18]
La IFN-α es otro producto de exposición prolongada a SAg. Esta citocina está estrechamente relacionada con la inducción de autoinmunidad,[19] y la enfermedad autoinmune La enfermedad de Kawasaki se sabe que es causada por una infección por SAg.[12]
La activación de SAg en células T conduce a la producción de ligando CD40 que activa el cambio de isotipo en células B a IgG e IgM e IgE.[20]
Para resumir, las células T son estimuladas y producen cantidades excesivas de citoquinas, lo que resulta en la supresión de las células T mediada por citocinas y la eliminación de las células activadas a medida que el cuerpo vuelve a la homeostasis. Los efectos tóxicos del microbio y el SAg también dañan los sistemas de tejidos y órganos, una condición conocida como síndrome de shock tóxico.[20]
Si se sobrevive la inflamación inicial, las células huésped se vuelven anérgicas o se eliminan, lo que resulta en un sistema inmunitario gravemente comprometido.
Efectos independientes (indirectos) de superantigenicidad
Además de su actividad mitogénica, los SAG pueden causar síntomas que son característicos de la infección.[1]
Uno de esos efectos es el vómito. Este efecto se siente en casos de intoxicación alimentaria, cuando las bacterias productoras de SAg liberan la toxina, que es altamente resistente al calor. Hay una región distinta de la molécula que es activa en la inducción de toxicidad gastrointestinal .[1] Esta actividad también es muy potente, y cantidades tan pequeñas como 20-35 μg de SAg pueden inducir el vómito.[8]
Los SAG pueden estimular el reclutamiento de neutrófilos en el sitio de infección de una manera que es independiente de la estimulación de células T. Este efecto se debe a la capacidad de los SAgs para activar las células monocíticas, estimulando la liberación de la citocina TNF-α, lo que lleva a una mayor expresión de moléculas de adhesión que reclutan leucocitos a las regiones infectadas. Esto causa inflamación en los pulmones, el tejido intestinal y cualquier lugar donde la bacteria haya colonizado.[21] Si bien pequeñas cantidades de inflamación son naturales y útiles, la inflamación excesiva puede conducir a la destrucción del tejido.
Uno de los efectos indirectos más peligrosos de la infección por SAg se refiere a la capacidad de los SAgs para aumentar los efectos de las endotoxinas en el cuerpo. Esto se logra reduciendo el umbral de endotoxicidad. Schlievert demostró que, cuando se administra de forma conjunta, los efectos de SAg y endotoxina aumentan hasta 50,000 veces.[7] Esto podría deberse a la eficiencia reducida del sistema inmunitario inducida por la infección por SAg. Además de la relación sinérgica entre la endotoxina y el SAg, el efecto de "doble golpe" de la actividad de la endotoxina y el SAg produce efectos más nocivos que los observados en una infección bacteriana típica. Esto también implica SAG en la progresión de la sepsis en pacientes con infecciones bacterianas.[20]
Enfermedades asociadas con la producción de superantígenos
Tratamiento
Los objetivos principales del tratamiento médico son estabilizar hemodinámicamente al paciente y, si está presente, eliminar el microbio que produce los SAG. Esto se logra mediante el uso de vasopresores, reanimación con líquidos y antibióticos.[1]
El cuerpo produce naturalmente anticuerpos contra algunos SAG, y este efecto puede aumentarse estimulando la producción de estos anticuerpos en las células B.[24]
Los grupos de inmunoglobulinas pueden neutralizar anticuerpos específicos y prevenir la activación de las células T. Se han creado anticuerpos y péptidos sintéticos para imitar regiones de unión a SAg en el MHC de clase II, bloqueando la interacción y evitando la activación de las células T.[1]
Los inmunosupresores también se emplean para prevenir la activación de las células T y la liberación de citocinas. Los corticosteroides se usan para reducir los efectos inflamatorios.[20]
Evolución de la producción de superantígenos
La producción de SAg efectivamente corrompe la respuesta inmune, permitiendo que el microbio que secreta el SAg sea transportado y transmitido sin control. Un mecanismo por el cual se hace esto es induciendo la anergia de las células T a antígenos y SAG.[13][16] Lussow y MacDonald demostraron esto al exponer sistemáticamente a los animales a un antígeno estreptocócico. Descubrieron que la exposición a otros antígenos después de la infección por SAg no logró provocar una respuesta inmune. En otro experimento, Watson y Lee descubrieron que las células T de memoria creadas por la estimulación de antígeno normal eran anérgicas a la estimulación de SAg y que las células T de memoria creadas después de una infección por SAg eran anérgicas a toda estimulación de antígeno. El mecanismo por el cual esto ocurrió fue indeterminado. Los genes que regulan la expresión de SAg también regulan los mecanismos de evasión inmune, como la proteína M y la expresión de la cápsula bacteriana, respaldando la hipótesis de que la producción de SAg evolucionó principalmente como un mecanismo de evasión inmune.[25]
Cuando la estructura de los dominios SAg individuales se ha comparado con otras proteínas estreptocócicas de unión a inmunoglobulinas (como las toxinas producidas por E. coli ), se encontró que los dominios se parecen por separado a los miembros de estas familias. Esta homología sugiere que los SAgs evolucionaron a través de la recombinación de dos motivos de cadena β más pequeños.[26]
SAG endógenos
Las exotoxinas estimulantes de linfocitos menores (Mls) se descubrieron originalmente en las células del estroma tímico de los ratones. Estas toxinas están codificadas por genes SAg que se incorporaron al genoma del ratón a partir del virus del tumor mamario del ratón (MMTV). La presencia de estos genes en el genoma del ratón permite que el ratón exprese el antígeno en el timo como un medio de seleccionar negativamente linfocitos con una región Beta variable que es susceptible a la estimulación por el SAg viral. El resultado es que estos ratones son inmunes a la infección por el virus más adelante en la vida.[1]
Aún no se ha identificado una selección dependiente de SAg endógena similar en el genoma humano, pero se han descubierto SAgs endógenos y se sospecha que desempeñan un papel integral en la infección viral. Se sabe que la infección por el virus de Epstein-Barr, por ejemplo, causa la producción de un SAg en las células infectadas, sin embargo, no se ha encontrado ningún gen para la toxina en el genoma del virus. El virus manipula la célula infectada para expresar sus propios genes SAg, y esto le ayuda a evadir el sistema inmunitario del huésped. Se han encontrado resultados similares con rabia, citomegalovirus y VIH.[1]
Referencias
- «Superantigens: microbial agents that corrupt immunity». Lancet Infect Dis 2 (3): 156-62. March 2002. PMID 11944185. doi:10.1016/S1473-3099(02)00222-0.
- Li H., Llera A., Malchiodi E.L., Mariuzza R.A. The structural basis of T cell activation by superantigens. Annu. Rev. Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435.
- «Streptococcus pyogenes: Insight into the function of the streptococcal superantigens». Int. J. Biochem. Cell Biol. 39 (1): 12-19. 2007. PMID 17029999. doi:10.1016/j.biocel.2006.08.009.
- «Interplay between superantigens and immunoreceptors». Scand. J. Immunol. 59 (4): 345-55. April 2004. PMID 15049778. doi:10.1111/j.0300-9475.2004.01404.x.
- «Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression». J. Exp. Med. 182 (5): 1573-7. November 1995. PMC 2192187. PMID 7595227. doi:10.1084/jem.182.5.1573.
- «Crystal structure of microbial superantigen staphylococcal enterotoxin B at 1.5 A resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors». J. Mol. Biol. 277 (1): 61-79. March 1998. PMID 9514739. doi:10.1006/jmbi.1997.1577.
- Schlievert PM (April 1982). «Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C». Infect. Immun. 36 (1): 123-8. PMC 351193. PMID 7042568. doi:10.1128/IAI.36.1.123-128.1982.
- «Staphylococcal and streptococcal superantigens: molecular, biological and clinical aspects». Int. J. Med. Microbiol. 292 (7–8): 429-40. February 2003. PMID 12635926. doi:10.1078/1438-4221-00232.
- «Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by group V superantigens». J. Mol. Biol. 367 (4): 925-34. April 2007. PMID 17303163. doi:10.1016/j.jmb.2007.01.024.
- «Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1». J. Mol. Biol. 353 (2): 308-21. October 2005. PMID 16171815. doi:10.1016/j.jmb.2005.08.041.
- «Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B». Immunity 9 (6): 807-16. December 1998. PMID 9881971. doi:10.1016/S1074-7613(00)80646-9.
- «Conservation and variation in superantigen structure and activity highlighted by the three-dimensional structures of two new superantigens from Streptococcus pyogenes». J. Mol. Biol. 299 (1): 157-68. May 2000. PMID 10860729. doi:10.1006/jmbi.2000.3725.
- «Defective T cell Receptor-mediated Signal Transduction in Memory CD4 T Lymphocytes Exposed to Superantigen or anti-T cell Receptor Antibodies». Cell. Immunol. 242 (2): 80-90. August 2006. PMC 1829409. PMID 17083922. doi:10.1016/j.cellimm.2006.09.008.
- «Molecular mechanisms for adaptive tolerance and other T cell anergy models». Semin. Immunol. 19 (3): 140-52. June 2007. PMC 2045643. PMID 17400472. doi:10.1016/j.smim.2007.02.005.
- Stiles BG, Krakauer (2005). «Staphylococcal Enterotoxins: a Purging Experience in Review, Part I». Clinical Microbiology Newsletter 27 (23): 23. doi:10.1016/j.clinmicnews.2005.11.001.
- «Differential effects of superantigen-induced "anergy" on priming and effector stages of a T cell-dependent antibody response». Eur. J. Immunol. 24 (2): 445-9. February 1994. PMID 8299694. doi:10.1002/eji.1830240227.
- «Anergy and Cytokine-Mediated Suppression as Distinct Superantigen-Induced Tolerance Mechanisms in Vivo». J. Exp. Med. 190 (1): 53-64. July 1999. PMC 2195559. PMID 10429670. doi:10.1084/jem.190.1.53.
- «Induction of negative regulators of haematopoiesis in human bone marrow cells by HLA-DR cross-linking». Transpl. Immunol. 7 (3): 159-68. September 1999. PMID 10608299. doi:10.1016/S0966-3274(99)80035-5.
- «Interferon-alpha-induced endogenous superantigen. a model linking environment and autoimmunity». Immunity 15 (4): 591-601. October 2001. PMID 11672541. doi:10.1016/S1074-7613(01)00212-6.
- «The superantigen toxic shock syndrome toxin-1 induces CD40 ligand expression and modulates IgE isotype switching». Int. Immunol. 8 (10): 1503-10. October 1996. PMID 8921429. doi:10.1093/intimm/8.10.1503.
- «Induction of acute inflammation in vivo by staphylococcal superantigens I: Leukocyte recruitment occurs independently of T lymphocytes and major histocompatibility complex Class II molecules». Lab. Invest. 78 (6): 647-56. June 1998. PMID 9645755.
- «New insights into the pathology of nasal polyposis: the role of superantigens and IgE». Verh K Acad Geneeskd Belg. 67 (5–28): 5-28; discussion 29-32. 2005. PMID 15828304.
- Salgado-Pabón W, et al. (2013) Superantigens are critical for Staphylococcus aureus infective endocarditis, sepsis, and acute kidney injury. MBio 4:e00494-00413.
- «Identification of the Antigenic Epitopes in Staphylococcal Enterotoxins A and E and Design of a Superantigen for Human Cancer Therapy». J. Mol. Biol. 333 (5): 893-905. 2003. PMID 14583188. doi:10.1016/j.jmb.2003.09.009.
- «High-frequency intracellular infection and erythrogenic toxin A expression undergo phase variation in M1 group A streptococci». Mol. Microbiol. 28 (1): 157-67. April 1998. PMID 9593304. doi:10.1046/j.1365-2958.1998.00786.x.
- «Staphylococcus aureus enterotoxins: a key in airway disease?». Allergy 57 (6): 480-7. June 2002. PMID 12028112. doi:10.1034/j.1398-9995.2002.02156.x.
Otras lecturas
- Superantigen Web Database en Birkbeck, Universidad de Londres
- Lista de proteínas de superantígeno Archivado el 18 de septiembre de 2019 en Wayback Machine. de UniProt
- MeSH: Superantigens (en inglés)
- Rasooly, R., Do, P. y Hernlem, B. (2011) Auto-presentación de la enterotoxina estafilocócica A por las células T CD4 + de ratón. Open Journal of Immunology, 1, 8-14.
Enlaces externos
 Wikimedia Commons alberga una categoría multimedia sobre Superantígeno.
Wikimedia Commons alberga una categoría multimedia sobre Superantígeno.
 Datos: Q772950
Datos: Q772950- Multimedia: Superantigens / Q772950