Groupe sanguin
Un groupe sanguin est une classification reposant sur la présence ou l'absence de substances antigéniques héritées à la surface des globules rouges (hématies). Ces antigènes peuvent être des protéines, des glucides, des glycoprotéines ou des glycolipides, selon le système de groupe sanguin, et certains de ces antigènes sont également présents à la surface d'autres types de cellules de différents tissus.
Pour le groupe humoristique québécois, voir Le Groupe sanguin.
Les divers groupes sanguins sont regroupés en systèmes. Appartient à un même système de groupes sanguins l'ensemble des épitopes ou phénotypes résultant de l'action des divers allèles d'un même gène ou de gènes étroitement liés.
Le sang est un tissu liquide que l’on peut facilement prélever sur un individu sain pour le transfuser à un individu malade. Or, malgré une composition cellulaire identique de ce tissu, il existe une variabilité, ou polymorphisme des divers éléments du sang entre les individus, ce qui rend impossible la transfusion entre certains groupes de personnes. On dit des personnes qui présentent une même caractéristique qu’elles appartiennent au même groupe sanguin. En règle générale, ces caractéristiques sont mises en évidence par des techniques d'hémagglutination grâce à des anticorps ou des lectines reconnaissant spécifiquement un épitope. En cas de problème, il peut être fait appel à la biologie moléculaire. Ces épitopes, déterminant divers phénotypes, sont génétiquement transmis.
La découverte du système ABO, le premier de ces systèmes, en 1900, par Karl Landsteiner[1] a permis de comprendre pourquoi certaines transfusions sanguines étaient couronnées de succès, alors que d'autres se terminaient tragiquement.
Bases d'immunologie
Les antigènes sont des molécules qui couvrent la surface de toutes les cellules de l'organisme et participent à son identité. Ils sont les cibles des anticorps lorsqu'ils sont identifiés comme étrangers. Mais les antigènes peuvent être aussi bien des substances extérieures à l'organisme et contre lesquelles réagissent les anticorps : le pollen, la poussière, certains aliments ou médicaments, ou les poils léchés d'animaux.
Les anticorps sont des molécules produites par les lymphocytes B du système immunitaire qui réagissent avec les antigènes n'appartenant pas à l'organisme. Ils attaquent le non-soi. Certains anticorps sont fabriqués « à la demande » (défense contre les bactéries…), d'autres existent naturellement dans l'organisme (ce qui fut découvert avec le système ABO).
Lorsqu'un anticorps (ou une lectine) se fixe spécifiquement à un antigène situé à la surface des globules rouges, il provoque l'agglutination, parfois l'hémolyse (destruction), de ces derniers. Cette agglutination peut être soit immédiate, et c'est ainsi que le système ABO a été découvert, soit « aidée » par une technique d'agglutination artificielle, et c'est ainsi, qu'après les travaux de Coombs, qui a produit et utilisé une antiglobuline, un grand nombre d'anticorps et de systèmes de groupes sanguins ont été découverts.
Historique de la découverte des groupes sanguins
La découverte des groupes sanguins est liée à la maîtrise de la pratique de la transfusion sanguine. Des essais de transfusions, souvent mortels pour le patient, ont été pratiqués avant le XIXe siècle en Occident. Jean Baptiste Denis a pratiqué sous Louis XIV la plus ancienne transfusion entièrement documentée connue et réussie le [2]. Une pratique sans compatibilité du sang est néanmoins dangereuse, à tel point que le parlement de Paris interdit cette pratique en 1668 malgré des réussites spectaculaires[2],[3].
La première découverte importante en Occident, après celle de la petite circulation sanguine (ou circulation pulmonaire) par Ibn Nafis à Damas en 1242, est celle de William Harvey en 1628, la circulation du sang, mais il faut attendre 1875 avec les travaux de l'Allemand Leonard Landois[4] et du Norvégien Jacob Worm-Müller (no)[5] pour voir que du sang humain mélangé à du sang animal s'agglutine en amas qui entraîne la mort du sujet transfusé. En 1900, Karl Landsteiner, médecin et biologiste autrichien, montre que le mélange de divers sangs humains peut aussi entraîner une agglutination. Il postule deux types de substances, les agglutinogènes sur les globules rouges et les agglutinines dans le sérum. Divers médecins et praticiens font avancer le sujet tels que Ludvig Hecktoen[6],[7], Werner Schultz (de)[8], James Blundell (en) et Alexis Carrel. En 1901, Karl Landsteiner découvre les groupes A, B et O tandis que ses élèves Alfred von Decastello et Adriano Sturli[9] découvrent le groupe AB en 1902[2]. Jan Janský définit en premier la classification ABO[10]. La transfusion devient saine après 1911 lorsque l'Américain Reuben Ottenberg (en) montre qu'il faut impérativement tenir compte des groupes d'isoagglutination.
Karl Landsteiner, en collaboration avec Philip Levine, découvre les groupes M, N et P en 1925[2]. En 1930, Landsteiner reçoit le prix Nobel de physiologie ou médecine pour ses travaux. Landsteiner, Alex Wiener, Philip Levine et Rufus E. Stetson découvrent le groupe Rhésus entre 1939 et 1940.
Classification
Ces différences antigéniques entre les individus définissent les différents groupes sanguins et peuvent porter aussi bien sur les éléments figurés du sang, globules rouges, globules blancs, plaquettes, que sur les protéines circulantes, en particulier les immunoglobulines. Le terme groupe sanguin ayant été appliqué aux seuls groupes connus avant les années 1950, à savoir aux groupes érythrocytaires, et ce terme étant souvent compris et en règle générale utilisé de façon restrictive dans cette acception, ce sont ces derniers qui seront traités dans la suite du présent article. Enfin, historiquement, ce sont les transfusions d'érythrocytes qui ont posé des problèmes cliniques d'incompatibilité, les autres éléments du sang n'étant que peu impliqués dans des accidents transfusionnels immédiats d'origine immunologique.
Concernant chacun des autres systèmes de groupes, lire les articles traitant de façon plus détaillée chacune de ces questions, ou y faisant référence, comme simple polymorphisme ou allotypie.
Groupes sanguins leucocytaires
C'est en travaillant avec des anticorps anti-leucocytes, et en tentant d'identifier des groupes leucocytaires, que Jean Dausset a découvert le système HLA. Il s'agissait en fait des antigènes d'histocompatibilité présents sur toutes les cellules de l'organisme. Certains antigènes du système HLA sont parfois présents sur les érythrocytes et donnent une réaction positive lors de la recherche d'anticorps irréguliers. Il s'agit d'antigènes dénommés Bg, soit Bga pour HLA-B7, Bgb pour HLA-B17, et Bgc pour HLA-A28/A2.
Les leucocytes portent également des antigènes spécifiques, soit aux différentes catégories de lymphocytes, soit aux polynucléaires. Ces derniers portent divers antigènes regroupés en 5 systèmes, HNA1, HNA2, HNA3, HNA4 et HNA5 (HNA pour Human Neutrophil Alloantigen).
Les anticorps dirigés contre les globules blancs, susceptibles d'être contenus dans un plasma transfusé, qu'il s'agisse d'anti HLA ou d'anti HNA, peuvent induire un accident transfusionnel grave, le TRALI (transfusion related acute lung injury) qui consiste en une atteinte œdémateuse pulmonaire.
Voir aussi transfusion sanguine et incompatibilité fœto-maternelle.
Groupes sanguins plaquettaires
Il s'agit des systèmes HPA (Human Platelet Antigens), au nombre de 6 : HPA1, HPA2, HPA3, HPA4, HPA5, et HPA15, tels que définis par le Comité de nomenclature des plaquettes, PNC (Platelet Nomenclature Committee) créé en 2003 en association avec l'ISBT et l'ISTH (Société internationale de thrombose et hémostase). Le plus connu de ces systèmes étant le système HPA1, suivi du système HPA5, dont les anticorps anti HPA1-a et HPA5-b sont impliqués respectivement dans 80 % et 10 % des cas d'incompatibilités fœto-maternelles plaquettaires.
Un anticorps dans l'un de ces systèmes entraîne :
- en cas d'incompatibilité fœto-maternelle, une thrombopénie chez le fœtus et le nouveau-né, causant parfois des hémorragies intracrâniennes qui peuvent être graves ;
- chez l'adulte, une transfusion inefficace en cas de transfusion de plaquettes incompatibles. Cette transfusion peut être exceptionnellement suivie d'un purpura post-transfusionnel où sont non seulement détruites immédiatement les plaquettes transfusées, mais également, par un mécanisme discuté, les propres plaquettes du patient.
Groupes sériques
Il s'agit des groupes Am, Gm, Km des immunoglobulines A, G, et de la chaîne légère Kappa, ainsi que du groupe ISf (inhibiteur San Francisco, situé sur la chaîne lourde des IgG1). Ces systèmes, dont le premier a été découvert par Grubb et Laurell, sont déterminés grâce à une antiglobuline, par une technique d'inhibition d'agglutination.
Groupes érythrocytaires
Il s'agit des premiers groupes sanguins qui ont été découverts (ABO, MNS), et le terme « groupes sanguins », utilisé de façon isolée, désigne en règle générale et de façon restrictive les groupes érythrocytaires, sinon on utilise le terme groupe plaquettaire, leucocytaire, ou sérique.
Les groupes sanguins sont identifiés usuellement avec des anticorps (immuno-typage), mais d'autres examens sont utiles. Par exemple, la plupart des lectines agglutinent les érythrocytes, se liant aux antigènes de groupe sanguin.
Groupes sanguins (érythrocytaires)
Les principaux groupes sanguins sont ceux qui définissent les systèmes ABO, Rhésus et Kell, mais il en existe beaucoup d'autres. Ces trois systèmes sont les plus importants, en pratique. Le premier, ABO, car il entraîne un accident transfusionnel immédiat en cas de transfusion incompatible et, de ce fait, a été le premier découvert. Le second, Rhésus, car l'immunogénicité de deux de ses antigènes (D - RH1 et c - RH4 surtout) entraîne très fréquemment des immunisations sources d'accidents ultérieurs et d'incompatibilités fœto-maternelles. Le troisième système, Kell, car l'antigène Kell est très immunogène, moins cependant que l'antigène RH1, D, et donne de ce fait, mais moins fréquemment, les mêmes complications.
La détermination du groupe
Elle est essentielle avant une transfusion, notamment en médecine d'urgence. Les techniques de typage du groupe nécessitent encore du temps et un équipement coûteux.
Dans les trois systèmes en ABO (A, B, AB ou O), en Rhésus (+ ou -), ou en Kell (+ ou -) la détermination du groupe se base, comme pour tous les systèmes, sur les caractéristiques des antigènes présents à la surface des érythrocytes et, pour le système ABO, sur les anticorps présents dans le sang.
En 2017, des chercheurs ont proposé un nouveau test rapide et bon marché où une gouttelette de sang est posée au centre d'une puce à base de papier contenant des anticorps anti-A à gauche et des anticorps anti-B à droite[11]. Quand le sang est absorbé par le papier jusque vers les extrémités de la puce, il entre en contact avec des zones contenant des anticorps, et s'y agglutine ou non, selon les marqueurs qu'il porte ; là où les globules rouges s'agglutinent, seul le plasma passe en colorant la puce d'une manière différente. 30 secondes suffisent pour obtenir le résultat[11]. Et le prototype du test peut être modifié pour les types de sang rares. Ces tests pourraient être très utiles dans les régions aux ressources médicales limitées et en situations d'urgence[12].
Classification des groupes sanguins
On donne ici la liste des différents systèmes définis et référencés par l'ISBT en 2015, avec dans l'ordre leur numéro, leur dénomination initiale ou commune, leur dénomination abrégée (symbole) officielle ISBT et HGNC, la nature de l'épitope ou de l'élément qui le porte, la localisation chromosomique, et le lien vers la référence OMIM. Enfin, selon la nomenclature de l'ISBT, dans chaque système, un numéro à 3 chiffres est attribué à chaque spécificité antigénique. Ainsi, dans le système ABO (001) quatre spécificités sont référencées : A=001, B=002, AB=003, A1=004. Dans le système MNS (002), on arrive au numéro 048, et dans le RH, on arrive (sept numéros ayant toutefois disparu) au numéro 061…
| Classification des groupes sanguins | |||||
|---|---|---|---|---|---|
| N° | Dénomination initiale ou commune | Dénominations abrégées
ISBT / HGNC |
Nature de l'épitope ou de l'élément qui le porte | Localisation chromosomique | Réf. OMIM |
| 001 | ABO | ABO / ABO | ose (N-acétylgalactosamine, galactose) | 9q34.2* | (en) 110300 |
| 002 | MNS | MNS / GYPA - GYPB - (GYPE) | GPA / GPB (glycophorines A et B). Possible récepteur de Plasmodium falciparum.
Antigènes M et N sur la GPA (mutations M→N : ser1leu et gly5glu), S et s sur la GPB (mutation S→s : met29thr). Par ailleurs, les vingt-six premiers AA N-terminaux extramembranaires de la GPA-N et de la GPB sont identiques (LSTTEVAMHT STSSSVTKSY ISSQTN…). Existence incertaine de la GPE. Le déficit en GPA entraîne le rarissime phénotype En(a)-, un déficit en acide sialique, et une chute du potentiel zêta. |
4q31.21 | (en) 111300
(en) 111740 (en) 138590 |
| 003 | P | P1PK / A4GALT | α1,4galactose sur paragloboside
Antigène [P1] : [Galα1-4Galβ1-4GlNacβ1]-3Lactosylcéramide (N.B. : l'antigène P est rattaché au système 028 : Globoside.) |
22q13.2 | (en) 111400 |
| 004 | Rhésus | RH / RHD - RHCE | protéines RHD / RHCE | 1p36.11 | (en) 111680 (en) 111700 |
| 005 | Lutheran | LU / BCAM | IgSF (apparenté aux immunoglobulines)
Liason génétique avec FUT2 (Secréteur), FUT3 (Lewis) et fraction C3 du Complément. |
19q13.32 | (en) 111200 |
| 006 | Kell | KEL / KEL | glycoprotéine, endopeptidase. Reliée (Cys72) par un pont disulfure à la protéine Kx (Cys347).
Lié au système Cartwright : LOD score de 3,48 pour Θ = 0,28 et au gène de reconnaissance du phénylthiocarbamide (PTC) à 14 cM - moyenne estimée. |
7q34 | (en) 110900 |
| 007 | Lewis | LE / FUT3 | ose (fucose), 2 gènes participent au phénotype Lewis. | 19p13.3 | (en) 111100 |
| 008 | Duffy | FY / DARC | protéine (ECR ou récepteur de chimiokine, et des Plasmodium vivax et Plasmodium knowlesi) | 1q23.2 | (en) 110700 |
| 009 | Kidd | JK / SLC14A1 | protéine (transporteur d'urée) | 18q12.3 | (en) 111000 |
| 010 | Diégo | DI / SLC4A1 | glycoprotéine (bande 3, AE 1, ou échangeur d'anions) | 17q21.31* | (en) 110500 |
| 011 | Cartwright | YT / ACHE | protéine (AChE, acétylcholinestérase, fixée à la membrane par le GPI ou glycosylphosphatidylinositol).
Lié au système Kell : LOD score de 3,48 pour Θ = 0,28 |
7q22.1 | (en) 112100 |
| 012 | Xg | XG / XG | glycoprotéine | Xp22.3 | (en) 314700 |
| 013 | Scianna | SC / ERMAP | glycoprotéine | 1p34.2 | (en) 111750 |
| 014 | Dombrock | DO / ART4 | ADP-ribosyltransférase (fixée à la membrane par le GPI) | 12p12.3 | (en) 110600 |
| 015 | Colton | CO / AQP1 | aquaporine 1 | 7p14.1 | (en) 110450 |
| 016 | Landsteiner-Wiener | LW / ICAM4 | IgSF (apparenté aux immunoglobulines) | 19p13.2 | (en) 111250 |
| 017 | Chido/Rodgers | CH/RG / C4A - C4B | C4A C4B (fractions du complément)
99 % d'homologie entre les deux protéines |
6p21.3 | (en) 120810 (en) 120820 |
| 018 | Hh | H / FUT1 | ose (fucose) | 19q13.33 | (en) 211100 |
| 019 | Kx | XK / XK | glycoprotéine. Reliée (Cys347) par un pont disulfure à la protéine Kell (Cys72). | Xp21.1 | (en) 314850 |
| 020 | Gerbich | GE / GYPC | GPC / GPD (glycophorines C et D, la GPD résultant d'une GPC ayant une délétion). Récepteur de P. falciparum discuté. | 2q14.3 | (en) 110750 |
| 021 | Cromer | CROM / DAF | glycoprotéine (DAF ou CD55, régulatrice des fractions C3 et C5 du complément, liée à la membrane par un GPI) | 1q32.2 | (en) 125240 |
| 022 | Knops | KN / CR1 | glycoprotéine (CR1 ou CD35, capteur d'immun-complexes) | 1q32.2 | (en) 607486 |
| 023 | Indian | IN / CD44 | glycoprotéine (CD44 fonction d'adhésion, ligand acide hyaluronique )
Mutation IN1 ⇒ IN2 : Pro46Arg, fréquence géniques IN1 : 0,014, IN2 : 0,986 |
11p13 | (en) 609027 |
| 024 | OK | OK / BSG | glycoprotéine Basigine (CD147) Superfamille des immunoglobulines | 19p13.3 | (en) 111380 |
| 025 | RAPH | MER2 / CD151 | Tétraspanine CD151 (glycoprotéine transmembranaire) | 11p15.5 | (en) 179620 (en) 602243 |
| 026 | John Milton Hagen | JMH / SEMA7A | protéine (liée à la membrane par un GPI) | 15q24.1 | (en) 607961 |
| 027 | Ii | I / GCNT2 | poly-N-acétyllactosaminoglycane non ramifié (i) / ramifié (I) | 6p24.2 | (en) 110800 |
| 028 | Globoside | GLOB / B3GALNT1 | acétylgalactosaminyltransférase 1
Antigène [P] : [GalNAcβ1-3Galα1]-4Galβ1-4Glβ1-1Céramide Récepteur du parvovirus B19[13] Lactosylcéramide : Galβ1-4Glβ1-1Céramide, également substrat de P1. |
3q26.1 | (en) 603094 |
| 029 | GIL | GIL / AQP3 | aquaporine 3 | 9p13.3 | (en) 607457 |
| 030 | Rh-associated glycoprotein | RHAG / RHAG | Glycoprotéine, 409aa, 36 % homologie avec RHCED. Indispensable à l'expression de RH. | 6p12.3 | (en) 180297 |
| 031 | Forssmann | FORS / GBGT1 | Glycosyltransférase. Tranfère en α1-3 une N-acétyl-galactosamine sur le globoside (antigène P) | 9q34.2 | (en) 606074 |
| 032 | JR | JR / ABCG2 | Protéine, 16 exons, 655 AA, 72 000 daltons, 3 sites de N-glycosylation possibles, un probable. Six passages membranaires. Fréquence des Jra+ >99 %. | 4q22.1 | (en) 614490 |
| 033 | LAN | LAN / ABCB6 | Protéine, 19 exons, 842 AA, 80 000 daltons, 10 cystéines, 4 sites possibles de N-glycosylation. Rôle dans la synthèse de l'hème. Sujets Lan- : 1/20 000. | 2q36 | (en) 111600 |
| 034 | VEL | VEL /SMIM1 | Protéine de membrane de l'érythrocyte (small integral membrane protein) dont la délétion donne le phénotype Vel négatif. | 1p36.32 | (en) 615264 |
| 035 | CD59 | CD59 / CD59 | Protéine de 128 AA, 5 ponts disulfures, N- et O-glycosylée, fixée à la membrane par un GPI. Protectrice vis-à-vis de la fraction C9 du complément. | 11p13 | (en) 107271 |
Il existe, n'appartenant pas à ces 35 systèmes, d'autres antigènes présents sur les érythrocytes. Ces antigènes sont classés dans deux séries, et des collections.
La première, série numérotée 700, regroupe (en 2015) 17 antigènes de faible incidence, rencontrés chez moins de 1 % des individus.
La seconde, série numérotée 901, regroupe en 2015, 7 antigènes (Ata, Emm, AnWj, Sda, PEL, ABTI, et MAM) de haute incidence, rencontrés chez plus de 90 % des individus, et très souvent plus de 99 % pour la plupart d'entre eux. Deux antigènes, Junior et Langereis, dont les gènes sont maintenant localisés ont quitté cette série en 2012 pour devenir les systèmes 32 et 33. Cela permettra maintenant, par biologie moléculaire, de trouver plus facilement et rapidement des donneurs compatibles permettant de transfuser les patients Lan- et Jra-, porteurs des anticorps correspondant.
Les « collections » numérotées 205 Cost (2 antigènes, Csa et Csb), 208 Er (3 antigènes, Era et Erb et Er3), 210 innominée (antigènes Lec et Led), et 213 MNCHO (6 antigènes), comprennent plusieurs antigènes, parfois antithétiques, pour chacune d'elles. L'antigène public Vel (OMIM (en) 615264 ) vient d'être localisé (fin 2013) sur la protéine membranaire SMIM1 (OMIM (en) 615242 )dont le gène est situé en 1p36.32, sur le bras court du chromosome 1[14]. Cette collection a donc maintenant intégré les divers systèmes de groupes sanguins sous le No 034 de l'ISBT. La collection 207, Ii, a vu son antigène I intégré à la liste des groupes sanguins sous le numéros 027, I de l'ISBT. La collection 209 Globoside, comprenant initialement 3 antigènes P, PK et LKE s'est vue diminuée de son antigène no 001, P, devenu le système 028 GLOB[15]. La collection MNCHO, comporte six antigènes, Hu, M1, Tm, Can, Sext, Sj. Ces antigènes sont portés par la GPA (M ou N), mais sont dus à sa O-glycosylation (taux anormaux d'acide sialique et de N-acétylglucosamine) et non à sa séquence d'acides aminés. Ils sont donc dus à une glycosyltransférase mutée, et cette collection ne peut, de ce fait, appartenir au système MNS. L'antigène M1 est présent chez 20 à 25 % des sujets noirs. Cette glycosylation anormale serait protectrice vis-à-vis du plasmodium falciparum.
Les antigènes rares, qu'ils appartiennent à un système, comme l'antigène Vw du système MNS, (système 002, antigène 009) ou à la série 700 comme les antigènes Peters (700.018) ou Rasmussen (700.040) sont appelés antigènes privés, et les anticorps correspondants anti-privés.
Les antigènes fréquents, qu'ils appartiennent à un système, comme les antigènes RH46, (système 004, antigène 046) ou U (système 002, antigène 005), à une collection comme l'antigène Era (208.001) ou à la série 901, comme les antigènes August (Ata - 901.003), Emm (901.008) ou Sid (Sda - 901.011) sont appelés antigènes publics, et les anticorps correspondants anti-publics.
Un système à l'origine dénommé Bg (Bennett-Goodspeed) n'est plus considéré comme un système de groupe érythrocytaire, depuis que l'on sait que ses trois antigènes Bga, Bgb, et Bgc qui étaient mis en évidence sur les globules rouges correspondent en fait respectivement aux antigènes du système HLA : HLA-B7, HLA-B17 et HLA-A28/A2. D'autres antigènes HLA ont aussi été parfois détectés, HLA-A10, A9, B8, B12, B15, de façon très variable selon les individus, et parfois de façon transitoire pendant quelques mois ou quelques années. Ces anticorps anti-HLA, hors les problèmes qu'ils peuvent poser au laboratoire lors des identifications d'anticorps irréguliers (difficulté levée par traitement des hématies du panel par la chloroquine, ou par EDTA/glycine-HCl[16]) n'ont jamais été impliqués dans des maladies hémolytiques du nouveau-né et n'auraient entraîné qu'une réaction hémolytique transfusionnelle, d'ailleurs discutée[17].
ABO et RH, modèles de groupes sanguins érythrocytaires
Ces deux systèmes sont les plus importants, tant dans la pratique médicale (avec le système Kell), que pour leur intérêt historique, car ils ont fourni les bases génétiques, immunologiques pour toutes les études ultérieures des autres systèmes.
Système ABO
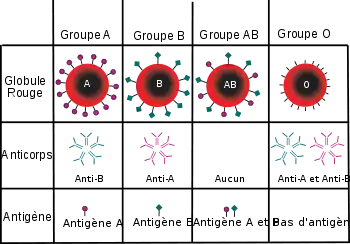
Découvert en 1900 par Landsteiner, le système ABO permet de classer les différents groupes sanguins selon la présence ou non d’antigènes A ou B à la surface des globules rouges.
Ainsi les globules rouges du groupe sanguin A possèdent des antigènes A, ceux du groupe B des antigènes B, ceux du groupe O aucun antigène, alors que ceux du groupe AB contiennent des antigènes de type A et de type B.
- La présence ou non d'anticorps anti-A ou anti-B dans le sérum. La présence d’antigènes d’un certain type impliquant l’absence d’anticorps de cette spécificité (sous peine de formation d’un complexe anticorps-antigènes !).
- Ces deux recherches d'antigènes définissant l'épreuve de Beth-Vincent, et d'anticorps définissant l'épreuve de Simonin-Michon sont obligatoires et doivent être concordantes pour établir un groupe sanguin ABO. Une exception toutefois chez le nouveau-né de moins de six mois dont les anticorps ne sont pas bien développés, et chez lequel ne sont donnés que des résultats non définitifs.
Système Rhésus
Ce système, expliquant certains problèmes indépendants du système ABO, accidents transfusionnels et la maladie hémolytique du nouveau-né dont la physiopathologie a été suspectée par Levine et Stetson en 1939, fut découvert et nommé en 1940 par Landsteiner et Wiener.
Le système Rhésus permet de classer les groupes sanguins selon la présence ou non d’antigène D à la surface des globules rouges (rhésus est le nom d'une espèce de macaque, Macaca rhesus, qui a permis de mettre en évidence ce système de groupe sanguin).
Dans la pratique médicale courante, on distingue les individus rh- qui ne portent pas l'antigène D, ou RH1 dans la nomenclature internationale, sur la surface de leurs hématies et les individus Rh+, qui présentent l'antigène D. En règle générale, les sujets rh- n'ont pas d'anticorps anti-D dans leur plasma. Une transfusion est alors possible sans conséquence immédiate.
Cet anticorps n'apparaît qu'après une transfusion non iso-rhésus (transfusion d'un sang D+, RH1, à un sujet D-) ou une grossesse après la naissance d'un enfant Rh+ chez une femme rh-. On dit alors qu'il s'agit d'un anticorps irrégulier. Dans ce dernier cas, la transfusion d'un sang Rhésus positif D+ entraîne une réaction hémolytique (qui détruit les hématies) par incompatibilité Rhésus.
Ce système de groupe sanguin comporte de nombreux autres antigènes à côté de l'antigène D = RH1. En particulier, les antigènes C (RH2), E (RH3), c (RH4) et e (RH5). Certains de ces antigènes peuvent entraîner les mêmes complications transfusionnelles ou fœtales que l'antigène D, en particulier l'antigène c (RH4), qui, lui, est en règle, présent chez un sujet rh négatif.
Répartition des groupes
En France, les groupes sanguins se répartissent de la manière suivante (exemple A+ prédomine avec 38 %) :
| Rhésus | Groupe sanguin | ||||
| O | A | B | AB | Total | |
| Rh+ | 36 % | 38 % | 8 % | 3 % | 85 % |
| Rh- | 6 % | 7 % | 1 % | 1 % | 15 % |
| Total : | 42 % | 45 % | 9 % | 4 % | 100 % |
Au Canada, en 2006 les groupes sanguins se répartissent de la manière suivante :
| Rhésus | Groupe sanguin | ||||
| O | A | B | AB | Total | |
| Rh+ | 39 % | 36 % | 7,5 % | 2,5 % | 85 % |
| Rh- | 7 % | 6 % | 1,5 % | 0,5 % | 15 % |
| Total : | 46 % | 42 % | 9 % | 3 % | 100 % |
Cette répartition peut présenter de sensibles différences selon les origines ethniques :
- chez les aborigènes d'Australie par exemple, on compte 68 % de O et 32 % de A ;
- chez les Inuits, 86 % de O ;
- chez les Asiatiques, on compte une plus forte proportion de groupe B.
Exemples de répartitions :
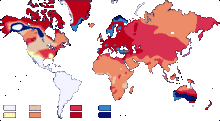
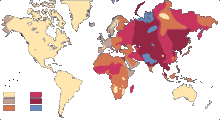
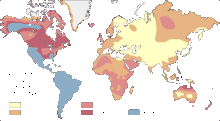
| Exemples de répartition dans le monde | |
| Groupe sanguin | Répartition mondiale |
|---|---|
| O + | 38 % |
| A + | 34 % |
| B + | 9 % |
| O - | 7 % |
| A - | 6 % |
| AB + | 3 % |
| B - | 2 % |
| AB - | 1 % |
| Exemples de répartition par type de population | ||||
| Population | O | A | B | AB |
| Allemande | 41 % | 43 % | 11 % | 5 % |
| Belge | 44 % | 45 % | 8 % | 3 % |
| Britannique | 47 % | 42 % | 8 % | 3 % |
| Basque | 56 % | 40 % | 3 % | 1 % |
| Autochtone du Pérou | 100 % | 0 % | 0 % | 0 % |
| Mayas | 97 % | 1 % | 1 % | 1 % |
| Autochtone d'Amérique | 96 % | 4 % | 0 % | 0 % |
| Oyirad (Russie) | 26 % | 23 % | 41 % | 11 % |
| Tchouvaches (près de Volga en Russie) | 30 % | 29 % | 33 % | 7 % |
De ces répartitions, on peut calculer les fréquences géniques dans les diverses populations, et donc les distances génétiques entre ces populations. Ce type de calculs est valable pour chaque système de groupe sanguin et pour chaque polymorphisme, par application de la loi de Castle-Hardy-Weinberg, article où le système ABO est pris pour exemple.
Compatibilité
La compatibilité entre le groupe sanguin d'un donneur et d'un receveur se pose lors des transfusions sanguines ou de transplantations. Une transfusion échouera si des anticorps rencontrent des cellules présentant les antigènes correspondants. Une réaction immunologique (agglutination et hémolyse) se déclencherait alors très rapidement pour détruire ces cellules. Les conséquences peuvent aller d'une transfusion inefficace sans signe clinique, à une réaction clinique légère (angoisse, frisson), grave (état de choc, hémoglobinurie, insuffisance rénale aiguë), ou dramatique (Choc, Coagulation intravasculaire disséminée) conduisant au décès.
Elle se pose également en cas de grossesse pour les femmes rhésus négatif portant un fœtus de rhésus positif. S'il s'agit d'une première grossesse, en général les choses se passent bien si la mère n'a pas été immunisée antérieurement par l'antigène D, RH1. Sinon, du fait que les anticorps peuvent franchir la barrière placentaire, les globules rouges du fœtus sont détruits plus ou moins massivement : c'est la maladie hémolytique du nouveau-né, ou MHNN. Cette maladie peut présenter tous les stades de gravité. Bénigne et n'entraîner qu'un simple ictère (jaunisse) et une anémie passagère, plus importante et nécessiter des transfusions, voire une exsanguino-transfusion à la naissance, majeure demandant un accouchement provoqué ou une césarienne avec exsanguino-transfusion immédiate, gravissime demandant des transfusions in utero pour éviter le décès de l'enfant, voire décès in utero de l'enfant avant toute intervention possible. Ces derniers cas sont devenus très rares depuis la prévention de l'immunisation des femmes par une injection d'anticorps anti-D vers la vingt-huitième semaine de grossesse (depuis 2005) puis à l'accouchement d'un enfant Rh Positif (depuis la fin des années 1960). Restent cependant les autres spécificités, dont les plus fréquentes sont l'anti-c (RH4), et l'anti-K1, qui causent également des maladies hémolytiques du nouveau-né.
Présence des anticorps
| Sang | Anticorps présents | |
| Anti-A | Anti-B | |
| Groupe O | ||
| Groupe A | ||
| Groupe B | ||
| Groupe AB | ||
Dans le système ABO, on retrouve dans le sang de toutes les personnes des anticorps spécifiques des antigènes qu'ils ne possèdent pas sur leurs globules. Ainsi une personne de groupe B développera naturellement des anticorps anti-A et une personne du groupe O développera des anticorps anti-A et des anticorps anti-B. Ces anticorps sont dits réguliers, car présents chez tous les individus, sauf chez le nouveau-né. Les anticorps anti-A par exemple se relient avec les molécules A sur les cellules.
Ces anticorps naturels apparaissent, dans le système ABO, dès les premiers mois de la vie. Ce sont des immunoglobulines de classe IgM, agglutinants et agissant à froid : ce sont des anticorps complets.
Dans le système Rhésus, il n'y a pas d'anticorps présents naturellement. Ils n'apparaissent en règle générale qu'après une première sensibilisation, par grossesse ou transfusion (certains tels l'anti-E ou l'anti-Cw pouvant toutefois être « naturels »). Ces anticorps apparus après sensibilisation sont dits irréguliers. Ce sont des immunoglobulines de classe IgG, actifs à 37 °C, et qui ne sont mis en évidence que par des techniques d'agglutination artificielle - techniques à l'antiglobuline ou aux enzymes. Ces anticorps, ne provoquant pas spontanément d'agglutination -mais pouvant provoquer une hémolyse in vivo- sont dits incomplets.
Transfusion de globules rouges
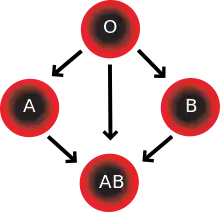
Les donneurs O peuvent donner aux receveurs O, A, B et AB ; les donneurs A peuvent donner aux receveurs A et AB ; les donneurs B peuvent donner aux receveurs B et AB ; les donneurs AB ne donnent qu'aux receveurs AB.
Les globules rouges (ou concentré érythrocytaire) sont extraits de dons de sang. En effet, le sang n'est plus que rarement transfusé dans son intégralité. Il est le plus souvent traité et séparé en ses composants.
Ainsi le concentré érythrocytaire ne contient que peu de plasma, donc peu d’anticorps. Les problèmes d’incompatibilité de la transfusion de plasma (voir ci-dessous) ne sont donc pas posés.
Lors d’une transfusion de globules rouges, il faut veiller à ne pas transmettre au receveur des cellules sanguines sur la surface desquelles se présentent des antigènes que le receveur n’a pas. Un traitement enzymatique, publié en 2007[19] et actuellement en expérimentation, permet d'éliminer les antigènes A et B des érythrocytes, et de les transformer en hématies de groupe O[20],[21]. Cette possibilité ne deviendra effective que dans quelques années, et, au début, pour des cas très particuliers de sangs dépourvus d'un antigène public présent chez plus de 99 % des donneurs, par exemple.
Les deux tableaux suivants doivent donc être respectés lors d'une transfusion :
|
| ||||||||||||||||||||||||||||||||||||||||
Ainsi, pour le système ABO, et en ne considérant que le groupe rhésus standard (antigène RH1), les sujets AB+ sont considérés comme receveurs universels, et les O- comme donneurs universels de globules rouges.
En ce qui concerne le système RH, cette règle est toujours considérée comme valable en cas d'urgence vitale, et en l'absence de groupe connu. Mais dès que l'on connaît le groupe du patient, il est souhaitable de respecter les autres antigènes de ce système, en particulier l'antigène c (RH 4), afin de ne pas immuniser les femmes jeunes en particulier.
Transfusion de plasma
Le plasma est un des composants du sang. Il est recueilli lors d'un don en plasmaphérèse et peut être utilisé (en France) pour transfusion directe, à condition d'avoir été prélevé chez un homme jamais transfusé (pour éviter la présence d'anticorps anti-HLA ou anti-HNA susceptibles d'induire un TRALI, et souvent présents chez les sujets transfusés et les femmes ayant eu des grossesses). Les plasmas extraits de dons de sang total ou par plasmaphérèses chez des femmes ne peuvent être utilisés (en France) que pour préparer des médicaments dérivés du sang, albumine, immunoglobulines, fibrinogène, fractions coagulantes, etc., ou des réactifs de laboratoire[pourquoi ?][réf. nécessaire].
Comme le plasma contient des anticorps en fonction de son groupe dans le système ABO, les globules rouges du receveur ne doivent pas présenter les antigènes correspondants. Le plasma de donneurs de groupe AB ne contenant pas d'anticorps convient à tous les receveurs.
Les plasmas contenant un anticorps, le plus souvent naturel dans un autre système (anti-P1, anti-Lewis, anti-M, etc.), ne sont pas utilisés en France. En ce qui concerne le système Rhésus, les plasmas contenant un anti-D (donc issus de femmes immunisées) étaient jusqu'à une époque relativement récente réservés, en France, pour la fabrication des immunoglobulines anti-D ou d'autres médicaments dérivés du sang. Ils ne le sont plus, (principe de précaution oblige) et les immunoglobulines anti-D utilisées pour la prévention de la maladie hémolytique du nouveau-né, issues de donneurs immunisés rémunérés, sont importées[réf. souhaitée]. Il est clair que, dans des pays où seraient utilisés des plasmas contenant un anti-D, le receveur ne pourrait être qu'un sujet rhésus négatif, tout comme le concentré érythrocytaire issu d'une donneuse ayant un anti-D ne pourrait être transfusé qu'à un patient Rh négatif.
En ce qui concerne leur immunogénicité, les plasmas frais congelés, ayant subi une inactivation virale S.D. (solvant-détergent) associée à une filtration ne sont pas immunogènes. Les plasmas qui étaient sécurisés par quarantaine (PFC-Se) et utilisés jusqu'en et qui sont à nouveau utilisés en remplacement du plasma viro-atténué au bleu de méthylène (PVA-BM) arrêté le , ou les plasmas viro-atténués par amotosalen, technologie Intercept (PVA-IA), peuvent contenir quelques hématies susceptibles, sinon de provoquer une immunisation primaire décelable, du moins de l'induire ou de relancer une immunisation secondaire[réf. souhaitée]. D'où la préférence qu'ont certains médecins de respecter pour ces plasmas (PFC-Se ou PVA-IA) une règle de compatibilité Rhésus identique à celle des globules rouges, sans que le bénéfice de cette attitude soit réellement documenté.
Les plasmas viro-atténués par méthode S.D. (solvant-détergent), ayant été ulta-filtrés, ne contiennent plus de stromas globulaires et ne peuvent être immunogènes. Pour ces plasmas, on ne tient pas compte du groupe Rhésus.
Le donneur et le receveur doivent donc respecter les tableaux suivants :
|
| ||||||||||||||||||||||||||||||||||||||||
Ainsi, alors que les personnes de groupe AB sont des receveurs universels de globules rouges dans le système ABO, ils sont donneurs universels de plasma. A contrario, si les personnes de groupe O (ne possédant pas les antigènes A ou B) sont des donneurs universels de globules rouges dans le système ABO, ils sont receveurs universels de plasma, possédant déjà les deux anticorps[22].
Impasses transfusionnelles
Certains malades posent d'énormes problèmes transfusionnels. Il s'agit en particulier de sujets dépourvus d'un antigène public, sujets Vel négatifs, RH:-46 ou KEL:-2, En(a)-, par exemple. On parle de sujets présentant un groupe sanguin rare ou phénotype érythrocytaire rare. En cas d'urgence vitale, ces personnes peuvent être transfusées une fois avec du sang classique si elles n'ont pas l'anticorps correspondant à leur groupe sanguin rare, mais ne peuvent plus l'être dès lors qu'elles sont immunisées, sauf par du sang rare identique au leur. De même certaines personnes ayant développé de nombreux anticorps ne peuvent recevoir que du sang d'un phénotype compatible dans les divers systèmes concernés, d'où leur rareté. Ces malades doivent donc dans la mesure du possible participer à un protocole d'auto-transfusion en cas d'intervention chirurgicale programmée, et faire si leur état de santé le permet des dons de sang qui seront conservés congelés à la Banque nationale des sangs de phénotype rare (BNSPR, Établissement français du sang Île-de-France, Créteil). Tous les patients et donneurs présentant un groupe sanguin rare sont suivis par le Centre national de référence pour les groupes sanguins (CNRGS, Paris), département de l'Institut national de la transfusion sanguine (INTS, Paris).
Génétique des groupes sanguins
Les groupes sanguins érythrocytaires sont définis grâce aux différences observées entre les individus à la surface des érythrocytes. Il s’agit donc de caractères allotypiques, c'est-à-dire différents d’un individu à l’autre à l’intérieur d’une même espèce. Ils sont déterminés par la paire de chromosome n°9
Ces différences portent sur la présence, l’absence ou l’agencement spatial à la surface des érythrocytes de sucres ou oses (systèmes ABO, P, etc.), ou de protéines (systèmes Rh, Kell, etc.). Autant de différences qui peuvent constituer un épitope antigénique pour quelqu’un qui ne le possède pas.
Ces caractères sont génétiquement transmis selon les lois de Mendel.
Système ABO
Caractérisé par deux sucres possibles à la surface de l’érythrocyte, soit un galactose (antigène B) soit une N-acétyl-galactosamine (antigène A). Ces sucres sont fixés sur une substance de base, appelée substance H, elle-même osidique. La présence de chacun de ces sucres est due à une enzyme spécifique codée par un gène lui-même spécifique : une variante A pour l’antigène A, B pour l’antigène B. La présence d’une enzyme inactive, due à un codon stop, pour ce gène ne permet pas l’ajout d’un sucre à cette substance de base H, qui reste donc en l’état. Cette enzyme inefficace, sans activité a été appelée « O » (O venant de ohne, « sans », en allemand), et a donné le groupe O.
Ainsi le système ABO est caractérisé par un gène dont il existe trois allèles (variantes du gène) A, B, et O. En réalité, il existe plusieurs variantes de l'allèle A, A1 et A2 en particulier, à l'origine de ces deux sous-groupes. Ce gène est porté par un autosome (par opposition aux chromosomes sexuels X ou Y). Tout individu possède donc deux copies du gène, l’un venant de son père et l’autre de sa mère, à un même locus, c’est-à-dire à un emplacement défini sur le chromosome. En l’occurrence, pour le système ABO, sur le chromosome 9.
Lorsque le sujet possède à la fois l'allèle A et le B, les deux sucres se trouvent alors sur l’érythrocyte et le sujet est de groupe AB. Lorsqu’il possède 2 allèles O, il sera de groupe O, s’il possède un ou deux A et pas l'allèle B, il sera A, s’il possède un ou deux allèles B et pas le A, il sera B.
Ainsi, un couple de parents, dont la mère est génétiquement A / O, donc de groupe A, et le père B / O, donc de groupe B pourra avoir des enfants de quatre groupes différents. Si chacun des parents transmet son allèle O, l’enfant sera génétiquement O / O, donc de groupe O. Si le père transmet l'allèle O et la mère le A, l’enfant sera A / O, donc de groupe A. Si le père transmet l'allèle B et la mère le O, l’enfant sera B / O, donc de groupe B. Si la mère transmet l'allèle A et le père celui B, l’enfant sera alors A / B, donc de groupe AB.
Système Rhésus
Il s’agit là d’un système de protéines. Deux gènes sont situés à des locus très proches l’un de l’autre sur le chromosome no 1, et sont donc transmis ensemble d’une génération à la suivante. Ces deux gènes résultent d’une duplication d’un gène originel, et synthétisent deux protéines très proches ayant la même structure et la même fonction ; si l’une est absente, l’autre la remplace, ce qui peut expliquer la grande quantité de protéines D chez les sujets ayant une délétion au locus CE (donc de phénotype D--, soit RH:1,-2,-3,-4,-5 en nomenclature internationale), ou les réactivités différentes des hématies selon le nombre de chacun des épitopes présents, lors d'une recherche d'anticorps irréguliers. Au premier locus, locus D, se trouve soit l’allèle D, qui synthétise la protéine Rhésus D définie par la présence de l’antigène D ou RH1, soit un emplacement vide dénommé d, qui ne synthétise rien. Au second locus, locus CE, se trouve un gène qui synthétise une seconde protéine qui ne porte pas l'épitope D. Mais cette seconde protéine présente deux autres épitopes principaux. L’un de ces épitopes définit les antigènes C ou c, le second les antigènes E ou e. La même protéine peut donc avoir quatre combinaisons possibles d'épitopes : ce, Ce, cE, CE.
Ainsi, en combinant l’ensemble de ces possibilités, on obtient huit agencements possibles, ou haplotypes, sur un même chromosome. Quatre de ces agencements comportent le gène D qui définira un sujet Rhésus positif standard. Il s’agit des haplotypes Dce, DCe, DcE, DCE. Quatre de ces agencements ne comportent pas le gène D. Il s’agit des haplotypes dce, dCe, dcE, dCE.
Le même raisonnement que pour les gènes du système ABO s’applique aux haplotypes du système Rhésus. Ainsi deux parents Rhésus positif de génotype D / d, donc hétérozygotes au locus D, pourront avoir un enfant rhésus négatif de génotype d / d.
Génétique des autres systèmes
L'ensemble des autres systèmes de groupes sanguins suit les mêmes lois génétiques. Cependant des particularités spécifiques à chacun des systèmes existent. Système Lewis par exemple, la synthèse de ses antigènes dépendant de deux systèmes génétiques (Lewis, avec ses allèles Le, le et système H avec ses allèles H, h), ou les systèmes Xg ou Kx dont les gènes sont situés sur le chromosome X, et non sur un chromosome autosomal
Anomalies apparentes et filiation
Dans chaque système de groupe sanguin, on peut être confronté à des anomalies apparentes de transmission.
Ainsi, dans le système ABO, l’antigène A résulte d’un sucre (ose) fixé par une enzyme sur une substance de base, également osidique, dite substance H. Cette même substance H résulte de l’action d’un gène H, que des très rares sujets ne possèdent pas. Ces sujets sont de génotype h/h, possédant en double dose l’allèle inactif h de H. Ces sujets sont dits de groupe « Bombay », du nom de la localité où cette particularité a été décrite. Ces sujets n’ont donc pas de substance H sur leurs globules rouges, et ont un anticorps anti-H dans leur plasma, ce qui interdit ou rend dangereuse toute transfusion non isogroupe (non « Bombay »). N’ayant pas cette substance H, même si ces sujets « Bombay » possèdent le gène A ou le gène B, les substances A ou B ne pourront être fabriquées, et ces sujets seront en apparence de groupe O. Leurs enfants héritant de ce parent d’un gène h et d’un gène A ou B, et de l’autre parent d’un gène normal H (dans le système Hh) et d’un gène O par exemple (dans le système ABO) pourront à nouveau exprimer le gène A ou B qui leur a été transmis par le premier parent et seront de groupe A ou B normal.
Le même problème peut se poser dans tout autre système où il existe un allèle amorphe, une délétion, une mutation ou un système inhibiteur. Il existe ainsi un rarissime haplotype rhnull dans le système Rhésus. Cet haplotype, qui ne synthétise aucune des deux protéines RH, ni RHD, ni RHCE, est noté RH:---. Supposons un père déterminé comme D+, C+, E-, c-, e+, c'est-à-dire possédant les antigènes D, C, et e, et ne possédant pas les antigènes c et E. On en déduit le génotype vraisemblable de ce père comme étant DCe / DCe, ou DCe / dCe. Or, ce père, uni à une femme de génotype dce / dce, pourra avoir un enfant D-, C-, E-, c+, e+, c'est-à-dire ne possédant pas l’antigène attendu C. Cet enfant sera considéré à tort comme de génotype dce/dce. On constate alors une apparente exclusion de paternité, l'enfant étant supposé avoir reçu un haplotype dce qui n'existe pas chez son père. Or ceci peut être parfaitement expliqué par le génotype DCe / --- de ce père, qui a transmis son haplotype « --- » à son enfant dont le génotype réel est dce / --- .
En conclusion, une anomalie apparente de transmission d’un groupe sanguin ne permet en aucune façon à elle seule de conclure à une exclusion de paternité ou de maternité. Une telle conclusion doit s’appuyer sur plusieurs systèmes, et maintenant sur la biologie moléculaire (analyse directe au niveau des chromosomes).
Anomalies, curiosités et pathologies
Difficultés de groupage
Les divers antigènes de groupes sanguins sont, en règle, mis en évidence par une technique d'agglutination, grâce à des anticorps ou des lectines. Dans certains cas d'anémies hémolytiques, ou après transfusion, il est impossible de déterminer le groupe sanguin du patient dans certains systèmes. Le phénotype dans les divers systèmes immunogènes (RH, KEL, FY, JK, MNS) est alors déduit du génotype obtenu en biologie moléculaire, par analyse directe de l'ADN. L'analyse des gènes permet également de définir et de classer les divers variants observés dans certains systèmes, en particulier RH, MNS, pour lesquels cette connaissance guide le choix de transfusions compatibles.
Antigènes faibles
Dans tous les systèmes, on peut voir des antigènes faibles, souvent signalés par un astérisque, ou un f -faible- en indice, ou un w -weak- en indice, sur les résultats du laboratoire, tels les A*, B*, E*, FY1*, KEL1f ou JK1w. Il est même parfois impossible de mettre ces antigènes en évidence par les techniques habituelles de groupage. Sont alors utilisées des techniques de fixation-élution, voire de biologie moléculaire si besoin.
Il en est ainsi des antigènes A faibles ou B faibles (A3, Ax, Am, etc., B3, Bx, etc.) pour lesquels c'est la faiblesse ou l'absence d'anticorps anti-A ou d'anti-B à l'épreuve de Simonin-Michon qui attire l'attention, et évite que ces groupes ne soient, à tort, étiquetés O. Cet antigène A, ou B, est cependant présent sur les érythrocytes, mais n'est pas mis, ou mal mis en évidence lors de l'épreuve globulaire de Beth-Vincent.
Dans le système RH, les antigènes D faibles sont encore appelés Du.
Tous les autres antigènes de groupe sanguin peuvent être affaiblis, pour diverses raisons, mutation du gène, manque de substrat, gène inhibiteur, etc. Ainsi, comme pour le Rhnull, existe un phénotype Lunull, donc Lu(a-, b-), dû soit à la présence d'un gène amorphe en double dose, cas où aucun antigène LU ne peut être mis en évidence, soit, le plus souvent, à l'action d'un gène inhibiteur. Il s'agit souvent d'un gène autosomique IN(Lu) actif en simple dose, cas où une très faible quantité d'antigène peut être mise en évidence sur les érythrocytes. Ce gène IN(Lu) provoque une forte dépression des antigènes Lutheran, para-Lutheran et AnWj (Anton), et un affaiblissement des antigènes de certains autres systèmes de groupes sanguins, P1, i, Indian, Knops. Existe également, dans quelques familles, un second gène inhibiteur Luthéran nommé XS2, lié au chromosome X, le gène normal étant nommé XS1, dont l'action est légèrement différente de In(Lu) sur les autres antigènes de groupe sanguin.
Certains antigènes de groupe sont connus pour donner, au laboratoire, des réactions très variables d'un individu à l'autre, tel l'antigène P1 chez l'adulte, ou donnent des réactions plus faibles chez le sujet hétérozygote que chez l'homozygote (effet de dose, antigènes M, N, S, Jka, etc.), ou ne sont pas développés à la naissance et apparaissent progressivement en l'espace de deux ou cinq ans, comme les antigènes Lewis ou P1.
Système Lewis
Certaines femmes Le(a-, b+) ou Le(a+, b-), pour 30 % d'entre elles, perdent pendant leur grossesse l'antigène Lewis qu'elles possèdent. Elles apparaissent donc comme Le(a-, b-) et développent un anticorps naturel anti-Lewis, anti-Lea, anti-Leb et/ou anti-Lex. Un mois au plus après l'accouchement, cet anticorps a disparu et ces femmes ont retrouvé leur phénotype Lewis normal. Cette perte d'antigène est sans conséquence pour l'érythrocyte, car la substance Lewis est une substance (glycosphingolipide) qui n'appartient pas à la membrane de l'érythrocyte, mais est une substance soluble (que l'on trouve dans le plasma, la salive, les larmes, le lait, le sperme, etc.) adsorbée passivement sur l'érythrocyte.
La substance Lewis n'est pas détectée sur l'érythrocyte du fœtus ni du nouveau-né qui est donc Le(a-, b-) à la naissance. Il apparaît Le(a+, b-) à l'âge d'un mois environ, puis Le(a+, b+) avant de devenir Le(a-, b+) vers l'âge de deux ans si tel doit être son phénotype définitif, lorsqu'il est génétiquement Le (gène Le) et Sécréteur (gène Se, référence OMIM (en) 182100 ), du moins chez les caucasiens. Ceci explique, entre autres raisons, que les anti-Lewis développés chez la mère n'ont aucune conséquence pour le fœtus.
Chimères hématopoïétiques
Lorsque les placentas de deux jumeaux dizygotes fusionnent et permettent une circulation croisée entre les fœtus, chacun d'entre eux possède alors ses propres cellules souches ainsi que celles de son jumeau ou de sa jumelle. Il y a greffe, tolérance immunitaire et les deux lignées cellulaires cohabitent chez le même individu. Dans chaque système, on peut observer une double population cellulaire due à la différence de groupe selon l'origine des cellules. Certains érythrocytes appartenant en propre à l'individu seront, par exemple, A, Rh+, K-, d'autres, venant de son jumeau pourront être B, rh-, K+. Parfois, en cas de mort précoce du second embryon, le chimérisme est une découverte fortuite pour l'individu survivant. C'est un cas qui peut poser problème dans les recherches en paternité, voire simuler une exclusion de maternité, le patrimoine génétique des cellules circulantes n'étant pas le même que celui des autres cellules somatiques ou germinales.
Parfois même il y a fusion précoce entre les deux œufs et il n'en résulte qu'un seul individu, ce qui ne pose pas de problème si les œufs sont du même sexe. Il en résulte un individu unique qui possède donc deux types de cellules, et pas seulement les cellules hématopoïétiques, chaque lignée cellulaire ayant son propre patrimoine génétique.
Les mêmes images de double population se voient régulièrement au laboratoire après transfusion, et dans le cas de greffes de moelle thérapeutiques. Cette double population est visible avant prise totale de la greffe, et réapparaît en cas de rejet.
Perte d'un antigène de groupe sanguin
Dans certaines affections préleucémiques, anémies réfractaires en particulier, certaines lignées d'érythrocytes peuvent ne plus synthétiser certains antigènes de groupes sanguins. Par exemple, un sujet connu de groupe AB, peut avoir trois types de globules en circulation, AB, A, et O, la première lignée n'étant pas atteinte, la seconde ayant perdu une enzyme, et la troisième en ayant perdu deux. Cette situation est appelée double population érythrocytaire. Ce constat est parfois un élément étiologique de l'anémie, bien avant les autres éléments cliniques.
Cette perte d'antigènes de groupes sanguins, qu'il s'agisse du système ABO ou d'un autre système, peut également s'accompagner d'une perte d'enzymes érythrocytaires (adélinate kinase), voire d'atteintes chromosomiques des autres lignées myéloïdes.
B acquis
Lors d'infections du tube digestif, lors de cancers coliques en particulier, certains germes libèrent une enzyme, une désacétylase, qui transforme la N-acétyl-galactosamine, qui constitue la substance A du groupe ABO, en galactosamine. Le phénotype B est alors acquis[23].
Lors d'un test sanguin, certains réactifs anti-B reconnaissaient aussi la galactosamine comme le galactose et réagissent dans ce cas comme si le groupe possédait la substance B. Le diagnostic pouvait alors être un groupe AB au lieu de A. Les réactifs commercialisés aujourd'hui sont à l'abri de cette réaction croisée[23].
Les réactifs maintenant commercialisés sont contrôlés et ne présentent plus, en principe, cette réaction croisée, qui pouvait être source d'erreur entre des mains inexpérimentées, faisant déterminer comme AB un sujet de groupe A.
Dès la fin de l'infection, l'anomalie disparaît progressivement.
Cis-AB et B(A)
Certaines mutations, Gly268Ala associée ou non à Arg176Gly sur la transférase A, font perdre à cette transférase sa spécificité et lui permet de transférer les deux oses (GalNac et Gal) sur la substance H si bien que le phénotype résultant d'un seul gène muté (en face d'un gène O) apparaît AB. La dénomination Cis-AB vient des études génétiques qui montraient que les deux spécificités enzymatiques sont codées en cis, c'est-à-dire sur le même chromosome, et non en trans comme attendu pour un groupe AB normal.
Deux autres mutations Pro234Ala ou Ser235Gly sur la transférase B altèrent également sa spécificité si bien que ces sujets B possèdent également un peu de substance A. Ce type de groupe est appelé B(A) ou B(A).
Autres pathologies ou curiosités
- Système Kell. Protéine Kell non exprimée chez le sujet de phénotype McLeod dont le gène XK, situé en Xp21.1, est génétiquement lié de façon étroite aux gènes de la rétinite pigmentaire (RP), de la granulomatose chronique (CGD) et de la myopathie de Duchenne (DMD), selon la séquence Xpter-DMD-XK-CGD-RP-Xcent. Une délétion à cet endroit expliquant la possible survenue de ces affections chez les sujets de phénotype McLeod, qui présentent par ailleurs une acanthocytose importante et une anémie hémolytique souvent bien compensée.
- Déficit en GPI (entraînant un déficit des protéines portant les systèmes YT, DO, JMH, CROM et l'antigène Emm-901.008). En particulier le déficit en protéine DAF / CD55 (decay-accelerating factor, antigènes Cromer), associé à un déficit en CD59 (MIRL membrane inhibitor of reactive lysis, glycoprotéine régulatrice du complément ne portant pas d'antigène de groupe sanguin) également liée par un GPI, sont à l'origine de l'hémoglobinurie paroxystique nocturne.
- Système I[24].
- Les autoanticorps anti-I ou anti Ii sont caractéristiques de la maladie des agglutinines froides. Ils apparaissent aussi de façon transitoire et à des titres inférieurs à la suite d'une infection respiratoire à mycoplasme (Mycoplasma pneumoniae). Ces anticorps ou auto anticorps (anti-I, HI, AI, BI) inactifs à 37 °C, d'où leur nom d'aggutinines froides, sont parfois présents à titre faible chez un sujet normal.
- Les sujets i, déficitaires en activité branchante I (GCNT2) par délétion ou mutation non sens (Gly348Glu ou Arg383His) du gène, sont atteints d'une cataracte congénitale, sans que le mécanisme physiopathologique en soit compris.
- À la naissance, les globules rouges du nouveau-né n'ont pas encore bien développé l'antigène I. On dit qu'ils sont de phénotype Ic, pour i de « cordon ».
- Enfin, les patients ayant une dysérythropoïèse (thalassémie, drépanocytose, Blackfan-Diamond, anémie réfractaire, HPN, etc.) ont une expression renforcée de l'antigène i. Une stimulation érythroïde pour compenser une anémie entraîne également une plus grande expression de i, l'enzyme branchante ayant eu moins de temps pour agir.
- Système RAPH. Certains sujets MER2 négatifs, ceux dont le gène de la protéine (en) 602243 présente un codon stop, sont atteints d'insuffisance rénale, de surdité et d'épidermolyse bulleuse prétibiale.
- RHnull et hémolyse chronique. Moins de 50 personnes dans le monde ont le « sang d’or »[25].
- Système Gerbich. Groupe GE négatif et elliptocytose.
- Systèmes FY, GE et résistance au paludisme. Les sujets FY:-1,-2 (grande fréquence en Afrique de l'Ouest et centrale[26] de 60 % - Soudan - à 100 % - Tanzanie, Zambie - selon les ethnies) étant protégés contre Plasmodiums vivax et knowlesi, les sujets GE négatifs (grande fréquence - 46,5 % - en Papouasie-Nouvelle-Guinée) étant, selon certaines études discutées, protégés contre Plasmodium falciparum.
- Système Globoside. L'anti P, hémolysine biphasique de Donath-Landsteiner, est cause de l'hémoglobinurie paroxystique a frigore. Proportion élevée d'avortements spontanés chez les femmes p [Tj(a-)] et p1k et p2k.
- Agammaglobulinémie et problème de détermination de groupe sanguin ABO - discordance entre les épreuves globulaires et sériques.
- Les taux de vWF et F VIII (facteurs de coagulation von Willebrand et antihémophilique A) sont plus faibles chez les sujets de groupe sanguin O. Les résultats d'un dosage doivent donc être interprétés en fonction du groupe sanguin.
Génétique des populations
Les fréquences géniques des allèles des groupes sanguins, calculées grâce à la loi de Castle-Hardy-Weinberg, ont permis l'essor de la génétique des populations. Grâce à elle, on peut suivre les migrations et les filiations des diverses populations du globe.
Notes et références
- (de) K. Landsteiner (1900), « Zur Kenntnis der antifermentativen, lytischen und agglutinierenden Wirkungen des Blutserums und der Lymphe » Zbl Bakt., 27, 357-362.
- (en) The History of Blood Transfusion Medicine, sur bloodbook.com
- La Transfusion Sanguine, sur donnersonsang.com
- (de) L. Landois, Die Transfusion des Blutes - Versuch einer physiologischen Begründung nach eigenen Experimental-Untersuchungen : mit Berücksichtigung der Geschichte, der Indicationen, der operativen Technik und der Statistik, 1875, F.C.W. Vogel, Leipzig.
- (de) Jacob Worm-Müller, Transfusion und Plethora : eine physiologische Studie, Christiana [Oslo], W. C. Fabritius, 1875.
- (en) Paul R. Cannon, Ludvig Hecktoen (1863-1951) A Biographical Memoir, NAS, (lire en ligne).
- (en) « Isoagglutination of Human Corpuscles with Respect to Demonstration of Opsonic Index and to Transfusion of Blood », Journal of the American Medical Association, vol. 48, p. 1739-1740.
- (de) Werner Schultz, « Ein weiterer Beitrag zur Transfusionsfrage », Berliner Klinische Wochenschrift, vol. 48, 1911, p. 34–36.
- (de) Alfred von Decastello et Adriano Sturli, « Ueber die Isoagglutinine im Serum gesunder und kranker Menschen », Münchener Medizinische Wochenschrift, vol. 49, 1902, p. 1090-1095.
- (cs) J. Janský, « Haematologick studie u. psychotiku », Sborn. Klinick, vol. 8, 1907, p. 85-139.
- Hong Zhang & al. (2017) A dye-assisted paper-based point-of-care assay for fast and reliable blood grouping ; Science Translational Medicine ; 15 mars 2017 ; Vol.9, n°381, eaaf9209 DOI: 10.1126/scitranslmed.aaf9209 (résumé)
- Lindzi Wessel (2017) Watch a special paper tool that can determine your blood type in seconds, Science Mag, News, publié 15 mars 2017
- (en) M.E. Reid, C. Lomas-Francis, The Blood Group Antigen (ISBN 978-0-12-586585-2)
- (en) BA Ballif, V Hélias, T Peyrard et al., « Disruption of SMIM1 causes the Vel- blood type », EMBO Mol. Med. 2013; 5: 751-761.
- (en) Human Blood Groups, Geoff Daniels, 3e éd., Wiley-Blackwell, 2013
- (en) K. Champagne, P. Sruell, J. Chen, L. Voll, G. Schlanser, « EDTA/glycine-acid versus chloroquine diphosphate treatement for stripping Bg antigens from red blood cells », Immunohematology 1999; 15:66-8.
- (en) G.E. Latoni, K Benson, G.F. Leparc, S. Agosti, « Hemolytic transfusion reaction due to autoantibody with HLA specificity », Transfusion 1999, 39-42S.
- Bloodbook
- (en) Qiyong P. Liu, Gerlind Sulzenbacher, Huaiping Yuan, Eric P. Bennett, Greg Pietz, Kristen Saunders, Jean Spence, Edward Nudelman, Steven B. Levery, Thayer White, John M. Neveu, William S. Lane, Yves Bourne, Martin L. Olsson, Bernard Henrissat, Henrik Clausen, « Bacterial glycosidases for the production of universal red blood cells », Nature Biotechnology, vol. 25, no 4, , p. 454-64 (ISSN 1087-0156, PMID 17401360, DOI 10.1038/nbt1298, lire en ligne, consulté le )
- (en) Martin L Olsson, Cheryl A Hill, Humberto de la Vega, Qiyong P Liu, Mark R Stroud, Jean Valdinocci, Steven Moon, Henrik Clausen, Margot S Kruskall, « Universal red blood cells--enzymatic conversion of blood group A and B antigens », Transfusion clinique et biologique: journal de la Société française de transfusion sanguine, vol. 11, no 1, , p. 33-9 (ISSN 1246-7820, PMID 14980547, DOI 10.1016/j.tracli.2003.12.002)
- (zh) Feng Gong, Qiu-Shuang Lü, Ying You, Hong-Wei Gao, Guo-Qiang Bao, Xin Gao, Su-Bo Li, Li-Li Li, Ying-Li Wang, Shu-Guang Tian, Zhi-Xin Zhang, Ping Zhang, Yang-Pei Zhang, « [Preparation of transfusable human universal red blood cell with recombinant alpha-galactosidase] », Zhongguo shi yan xue ye xue za zhi / Zhongguo bing li sheng li xue hui = Journal of experimental hematology / Chinese Association of Pathophysiology, vol. 13, no 2, , p. 313-6 (ISSN 1009-2137, PMID 15854299)
- AFSSaPS : Publications, Bonnes Pratiques, Transfusion
- Jacques Chiaroni, Pascal Bailly et Francis Roubinet, Les groupes sanguins érythrocytaires, La Plaine Saint-Denis, Établissement français du sang, (OCLC 908234222), p. 9
- Human Blood Groups, Geoff Daniels, 2e éd., 2002 Blackwell Science
- « Owdin.live : Golden blood : le sang le plus rare au monde », sur OWDIN, (consulté le )
- Hassan et al. 1968, Barnicot et al. 1968, Barclay et al. 1969. Dans The distribution of Human Blood Groups and other polymorphisms, A.E. Mourant et al., Oxford Medical Publication 1976.
Voir aussi
Bibliographie
- (en) Human blood groups, Geoff Daniels, Blackwell Science Ltd, 3e éd., 2013.
- (en) The blood group antigen, Marion E. Reid, Christine Lomas-Francis et Martin L. Olsson, Facts Book, Elsevier Academic Press, 3e éd., 2012.
- (en) Nomenclature of human platelet antigens, Vox Sanguinis 2003;85:240-245
- Les analyses immunohématologiques et leurs applications cliniques, J. Chiaroni, F. Roubinet, P. Bailly, L. Mannessier, F. Noizat-Pirenne, Édit. : John Libbey Eurotext, 2011
- Les groupes sanguins érythrocytaires, coordonné par P. Bailly, J. Chiaroni, F. Roubinet, Édit. : John Libbey Eurotext, Paris, 2015.
Articles connexes
Liens externes
- (en) (ISBT)
- (en) Liste des systèmes référencés à l'ISBT, ISBT
- (en) Nomenclature des groups sanguins, ISBT
- (en) National Center for Biotechnology Information (NCBI)
- (en) HUGO Nomenclatue Committee (HGNC) - groupes sanguins
- Groupes sanguins rares, par T. Peyrard, CNRGS
 Portail de la médecine
Portail de la médecine  Portail de la biologie
Portail de la biologie  Portail de l’hématologie
Portail de l’hématologie