Glycogen storage disease type IV
| Glycogen storage disease type IV | |
|---|---|
| Other names: Andersen's triad, Andersen’s disease[1], Glycogenosis type IV, Glycogen branching enzyme deficiency, Polyglucosan body disease, Amylopectinosis | |
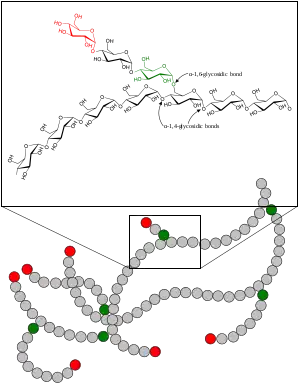 | |
| Glycogen | |
Glycogen storage disease type IV (GSD IV), or Andersen's Disease,[2][3] is a form of glycogen storage disease, which is caused by an inborn error of metabolism. It is the result of a mutation in the GBE1 gene, which causes a defect in the glycogen branching enzyme. Therefore, glycogen is not made properly and abnormal glycogen molecules accumulate in cells; most severely in cardiac and muscle cells. The severity of this disease varies on the amount of enzyme produced. GSD IV is autosomal recessive, which means each parent has a mutant copy of the gene, but show no symptoms of the disease. Having an autosomal recessive inheritance pattern, males and females are equally likely to be affected by Andersen's disease. Classic Andersen's disease typically becomes apparent during the first few months after the patient is born. Approximately 1 in 20,000 to 25,000 newborns have a glycogen storage disease.[4] Andersen's disease affects 1 in 800,000 individuals worldwide, with 3% of all GSDs being type IV.[5] The disease was described and studied first by Dorothy Hansine Andersen.[6][7]
Mutations in GBE1 can also cause a milder disease in adults that is called adult polyglucosan body disease.[8]
Variant types
Fatal perinatal neuromuscular type
- Excess fluid builds up around and in the body of the fetus
- Fetuses exhibit fetal akinesia deformation sequence
- Causes decrease in fetal movement and stiffness of joints after birth
- Infants have low muscle tone and muscle wasting
- Do not survive past the newborn stage due to weakened heart and lungs
Congenital muscular type
- Develops in early infancy
- Babies have dilated cardiomyopathy, preventing the heart from pumping efficiently
- Only survive a few months
Progressive hepatic type
- Infants have difficulty gaining weight
- Develop enlarged liver and cirrhosis that is irreversible
- High BP in hepatic portal vein and buildup of fluid in the abdominal cavity
- Die of liver failure in early childhood
Non-progressive hepatic type
- Same as progressive, but liver disease is not so severe
- Do not usually develop cirrhosis
- Usually show muscle weakness and hypotonia
- Survive into adulthood
- Life expectancy varies upon symptom severity
Childhood neuromuscular type
- Develops in late childhood
- Has myopathy and dilated cardiomyopathy
- Varies greatly
- Some have mild muscle weakness
- Some have severe cardiomyopathy and die in early adulthood
Pathology
It is a result of the absence of the glycogen branching enzyme, which is critical in the production of glycogen. This leads to very long unbranched glucose chains being stored in glycogen. The long unbranched molecules have low solubility, leading to glycogen precipitation in the liver. These deposits subsequently build up in the body tissue, especially the heart and liver. The inability to break down glycogen in muscle cells causes muscle weakness. The probable result is cirrhosis and death within five years. In adults, the activity of the enzyme is higher and symptoms do not appear until later in life.
Diagnosis
An assay of amylo-1,4 → 1,6 glucan transferases (which removes a block of 6 glucose residues from the 1,4 position and attaches it to the 1,6 position of the same chain)
Differential diagnosis
The DDx for Glycogen storage disease type IV is as follows:[9]
- Spinal muscular atrophy
- Pompe disease
- Zellweger spectrum disorder
- Congenital disorders of glycosylation
Management
The treatment for Glycogen storage disease type IV is as follows:[9]
- Adequate dietary intake
- Physical therapy
- Liver transplant
- Cardiac transplant
In animals
The form in horses is known as glycogen branching enzyme deficiency. It has been reported in American Quarter Horses and related breeds.
The disease has been reported in the Norwegian Forest Cat, where it causes skeletal muscle, heart, and CNS degeneration in animals greater than five months old. It has not been associated with cirrhosis or liver failure.[10][11]
References
- ↑ "Andersen Disease (GSD IV)". Archived from the original on 2023-02-05. Retrieved 2023-02-05.
- ↑ Andersen's disease (Dorothy Hansine Andersen) at Who Named It?
- ↑ Andersen DH (1956). "Familial cirrhosis of the liver with storage of abnormal glycogen". Lab. Invest. 5 (1): 11–20. PMID 13279125.
- ↑ "Andersen Disease (GSD IV)". National Organization for Rare Disorders. Archived from the original on 5 February 2023. Retrieved 5 February 2023.
- ↑ "Glycogen Storage Disease Type IV." Genetics Home Reference. U.S. National Library of Medicine, 10 Sept. 2015. Web. 27 Sept. 2015.
- ↑ Marsden, Deborah. "Andersen Disease (GSD IV)". NORD (National Organization of Rare Diseases). Archived from the original on 5 February 2023. Retrieved 1 December 2018.
- ↑ Mosby, By (2010). Mosby's Pocket Dictionary of Medicine, Nursing & Health Professions - E-Book (6 ed.). St. Louis, Missouri: Mosby Inc. p. 74. ISBN 978-0-323-05291-7. Archived from the original on 29 October 2023. Retrieved 1 December 2018.
- ↑ McKusick, Victor A.; Kniffin, Cassandra L. (May 2, 2016). "OMIM Entry 263570 - Polyglucosan body neuropathy, adult form". Online Mendelian Inheritance in Man. Johns Hopkins University. Archived from the original on 5 October 2020. Retrieved 7 March 2017.
- 1 2 Magoulas, Pilar L.; El-Hattab, Ayman W. (1993). "Glycogen Storage Disease Type IV". GeneReviews®. University of Washington, Seattle. Archived from the original on 2023-02-02. Retrieved 2023-10-26.
- ↑ Fyfe, JC; Giger, U; Van Winkle, TJ; Haskins, ME; Steinberg, SA; Wang, P; Patterson, DF (December 1992). "Glycogen storage disease type IV: inherited deficiency of branching enzyme activity in cats". Pediatric Research. 32 (6): 719–25. doi:10.1203/00006450-199212000-00020. PMID 1337588.
- ↑ Fyfe, J. C.; Kurzhals, R. L.; Hawkins, M. G.; Wang, P.; Yuhki, N.; Giger, U.; Van Winkle, T. J.; Haskins, M. E.; Patterson, D. F.; Henthorn, P. S. (2007). "A complex rearrangement in GBE1 causes both perinatal hypoglycemic collapse and late-juvenile-onset neuromuscular degeneration in glycogen storage disease type IV of Norwegian forest cats". Molecular Genetics and Metabolism. 90 (4): 383–392. doi:10.1016/j.ymgme.2006.12.003. PMC 2063609. PMID 17257876. "Deficiency of glycogen branching enzyme (GBE) activity causes glycogen storage disease type IV (GSD IV), an autosomal recessive error of metabolism. Abnormal glycogen accumulates in myocytes, hepatocytes, and neurons, causing variably progressive, benign to lethal organ dysfunctions. A naturally occurring orthologue of human GSD IV was described previously in Norwegian Forest cats (NFC)."
External links
| Classification | |
|---|---|
| External resources |
|