Transaldolase deficiency
| Transaldolase deficiency | |
|---|---|
| The structure of the Tranaldolase enzyme.[1][2] | |
Transaldolase deficiency is a disease characterised by abnormally low levels of the transaldolase enzyme. It is a metabolic enzyme involved in the pentose phosphate pathway. It is caused by mutation in the transaldolase gene (TALDO1). It was first described by Verhoeven et al. in 2001.[3]
Signs and Symptoms
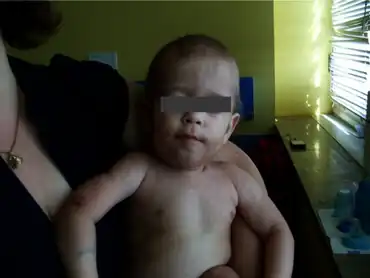
The leading symptoms of Transaldolase Deficiency are coagulopathy, thrombocytopenia, hepatosplenomegaly, hepatic fibrosis and dysmorphic features.[4] The dysmorphic features can include antimongoloid slant, low-set ears, and cutis laxa.[5] Those affected by this disease have abnormal polyol concentrations in urine and other bodily fluids, this can determined by an abnormal liver function tests.[4][5]
With transaldolase deficiency there is a buildup of sedoheptulose 7-phosphate (it is increased six to sevenfold in the blood compared to normal), which decreases the change of ribose 5-phosphate into glucose 6-phosphate.[6][7] This reaction is important in releasing NADPH. Reduced glutathione is essential for regulation of Mitochondrial membrane permeability and depends on the NADPH generated from the pentose phosphate pathway to be regenerated from oxidized glutathione.[8] Transaldolase plays an important role in male fertility; this is because it maintains the mitochondrial transmembrane potential and its role in the release NADPH. Therefore, transaldolase deficiency decreases the mobility of spermatozoa and lowers male fertility.[7]
Liver cirrhosis is associated with increased apoptosis of hepatocytes and transaldolase is a regulator in apoptosis signaling processing – therefore transaldolase deficiency can result in liver cirrhosis.[8]
Causes
TALDO1 is either mutated by the deletion of residue Ser171 or a replacement of Arg192 by His or Cys, changing the formation of the protein.[10] The deletion of Ser171 leads to inactivation and proteasome-mediated degradation of TALDO1.[11]
This shows the pentose phosphate pathway in humans with transaldolase catalysing the following reaction:[9]
D-glyceraldehyde 3-phosphate + D-sedoheptulose 7-phosphate <=> D-fructose 6-phosphate + D-erythrose 4-phosphate
Diagnosis
There are two different techniques for the diagnosis of Transaldolase deficiency.
Metabolite Analyses
Autozygome analysis and biochemical evaluations of urinary sugars and polyols can be used to diagnose Transaldolase Deficiency.[12] Two specific methods for measuring the urinary sugars and polyols are liquid chromatographytandem mass spectrometry and gas chromatography with flame ionization detection.[5]
Mutation Analysis
Direct sequence analysis of genomic DNA from blood can be used to perform a mutation analysis for the TALDO1 gene responsible for the Transaldolase enzyme.[5]
Treatment
At this time there is no treatment for transaldolase deficiency.[13]
There is currently research being done to find treatments for transaldolase deficiency. A study done in 2009 used orally administered N-acetylcysteine on transaldolase deficient mice and it prevented the symptoms associated with the disease.[14] N-acetylcysteine is a precursor for reduced glutathione, which is decreased in transaldolase deficient patients.[8][14]
Epidemiology
Transaldolase deficiency is recognized as a rare inherited pleiotropic metabolic disorder first recognized and described in 2001 that is autosomal recessive.[15][4][12] There have been only a few cases that have been noted, as of 2012 there have been 9 patients recognized with this disease and one fetus.[4]
See also
Transaldolase
References
- ↑ Thorell S, Gergely P, Banki K, Perl A, Schneider G (Jun 2000). "The three-dimensional structure of human transaldolase". FEBS Lett. 475 (3): 205–8. doi:10.1016/s0014-5793(00)01658-6. PMID 10869557. S2CID 33590067.
- ↑ Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (October 2004). "UCSF Chimera–a visualization system for exploratory research and analysis". J Comput Chem. 25 (13): 1605–12. doi:10.1002/jcc.20084. PMID 15264254. S2CID 8747218.
- ↑ Verhoeven NM, Huck JH, Roos B, et al. (May 2001). "Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway". Am. J. Hum. Genet. 68 (5): 1086–92. doi:10.1086/320108. PMC 1226089. PMID 11283793.
- 1 2 3 4 Loeffen YG, Biebuyck N, Wamelink MM, Jakobs C, Mulder MF, Tylki-Szymańska A, Fung CW, Valayannopoulos V, Bökenkamp A (Aug 2012). "Nephrological abnormalities in patients with transaldolase deficiency". Nephrol Dial Transplant. 27 (8): 3224–7. doi:10.1093/ndt/gfs061. PMID 22510381.
- 1 2 3 4 Balasubramaniam S, Wamelink MM, Ngu LH, Talib A, Salomons GS, Jakobs C, Keng WT (Jan 2011). "Novel heterozygous mutations in TALDO1 gene causing transaldolase deficiency and early infantile liver failure". J Pediatr Gastroenterol Nutr. 52 (1): 113–6. doi:10.1097/MPG.0b013e3181f50388. PMID 21119539. S2CID 7342726.
- ↑ Vas G, Conkrite K, Amidon W, Qian Y, Bánki K, Perl A (Apr 2006). "Study of transaldolase deficiency in urine samples by capillary LC-MS/MS". J Mass Spectrom. 41 (4): 463–9. Bibcode:2006JMSp...41..463V. doi:10.1002/jms.1004. PMC 3127395. PMID 16470722.
- 1 2 Perl A, Qian Y, Chohan KR, Shirley CR, Amidon W, Banerjee S, Middleton FA, Conkrite KL, Barcza M, Gonchoroff N, Suarez SS, Banki K (Oct 2006). "Transaldolase is essential for maintenance of the mitochondrial transmembrane potential and fertility of spermatozoa". Proc Natl Acad Sci U S A. 103 (40): 14813–8. Bibcode:2006PNAS..10314813P. doi:10.1073/pnas.0602678103. PMC 1595434. PMID 17003133.
- 1 2 3 Perl A (Jun 2007). "The pathogenesis of transaldolase deficiency". IUBMB Life. 59 (6): 365–73. doi:10.1080/15216540701387188. PMID 17613166.
- 1 2 Romero P.; Wagg J.; Green M.L.; Kaiser D.; Krummenacker M.; Karp P.D. (2004). "Computational prediction of human metabolic pathways from the complete human genome". Genome Biology. 6 (1): 1–17. doi:10.1186/gb-2004-6-1-r2. PMC 549063. PMID 15642094.
- ↑ Samland AK, Sprenger GA (Jul 2009). "Transaldolase: from biochemistry to human disease". Int J Biochem Cell Biol. 41 (7): 1482–94. doi:10.1016/j.biocel.2009.02.001. PMID 19401148.
- ↑ Wamelink MM, Struys EA, Jakobs C (Dec 2008). "The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review". J Inherit Metab Dis. 31 (6): 703–17. doi:10.1007/s10545-008-1015-6. PMID 18987987. S2CID 30263176.
- 1 2 Jassim N, Alghaihab M, Al Saleh S, Alfadhel M, Wamelink MM, Eyaid W (2013). "Pulmonary Manifestations in a Patient with Transaldolase Deficiency". JIMD Reports - Volume 12. JIMD Rep. JIMD Reports. Vol. 12. pp. 47–50. doi:10.1007/8904_2013_243. ISBN 978-3-319-03460-7. PMC 3897798. PMID 23846909.
- ↑ Wamelink MM, Struys EA, Salomons GS, Fowler D, Jakobs C, Clayton PT (Jun 2008). "Transaldolase deficiency in a two-year-old boy with cirrhosis". Mol Genet Metab. 94 (2): 255–8. doi:10.1016/j.ymgme.2008.01.011. PMID 18331807.
- 1 2 Hatting M.; Trautwein C.; Cubero F. J. (2009). "TAL deficiency, all roads lead to oxidative stress?". Hepatology. 50 (3): 979–981. doi:10.1002/hep.23227. PMID 19714731.
- ↑ Eyaid W, Al Harbi T, Anazi S, Wamelink MM, Jakobs C, Al Salammah M, Al Balwi M, Alfadhel M, Alkuraya FS (Nov 2013). "Transaldolase deficiency: report of 12 new cases and further delineation of the phenotype". J Inherit Metab Dis. 36 (6): 997–1004. doi:10.1007/s10545-012-9577-8. PMID 23315216. S2CID 25110878.
External links
| Classification | |
|---|---|
| External resources |
|