CYLD cutaneous syndrome
| CYLD cutaneous syndrome | |
|---|---|
| Other names: Multiple familial trichoepithelioma, Brooke–Spiegler syndrome, familial cylindromatosis, epithelioma adenoides cysticum | |
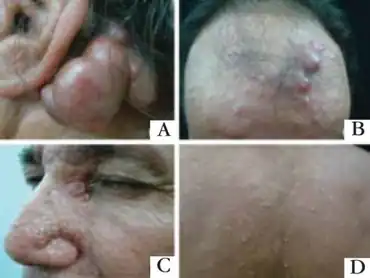 | |
| a)Multilobular violaceous nodule b) erythematous papules and nodules c) normochromic papules on eyelids d) normochromicpapules on the back | |
| Specialty | Dermatology, surgery, oncology |
| Symptoms | Multiple benign skin tumors |
| Complications | Malignant tumor formation |
| Usual onset | At puberty or earlier |
| Duration | Lifetime |
| Causes | CYLD gene mutations |
| Treatment | Surgical |
| Frequency | Rare |
| Deaths | Rare |
CYLD cutaneous syndrome (CCS) is the recently designated term for three rare inherited cutaneous adnexal tumor syndromes: multiple familial trichoepithelioma (MFT1) (also termed epithelioma adenoides cysticum and epithelioma adenoides cysticum of Brooke[1]), Brooke–Spiegler syndrome (BSS), and familial cylindromatosis (FC).[2] Cutaneous adnexal tumors are a large group of skin tumors that consist of tissues that have differentiated (i.e. matured from stem cells) towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands.[3] CCS tumors are hair follicle tumors.[2]
Individuals with the MFT1, BSS, and FC forms of CCS carry a germline (i.e. present in the germ cells which give rise to an individual) mutation in one of their two CYLD (i.e. CYLD lysine 63 deubiquitinase) genes. These individuals have skin tumors that tend to cluster into MFT1, BSS, and/or FC types that differ form each other in their locations, organizations, and microscopic appearances.[4] Nonetheless, members of a single family with CCS can manifest either a FC-, MFT1- or BSS-type pattern.[5] Furthermore, these different patterns have little or no impact on the prognoses or course of individuals with CCS.[4] The term “CYLD cutaneous syndrome” as applied to individuals with MFT1, BSS, or FC hair follicular tumors and carrying a hereditary CYLD mutation was first proposed by Rajan et al. in 2009.[6]
Individuals generally develop increasing numbers of benign skin tumors beginning in their youth and continuing throughout most of their lives. In uncommon cases, they develop malignant tumors many of which appear to arise from their benign tumors. Tumors that are unacceptably symptomatic, highly disfiguring, or malignant are treated by surgical excision methods plus, in cases of malignancy or other medial considerations, radiation therapy. Radiation therapy alone may be used in cases of surgically inaccessible tumors. All individuals with CCS should have routine yearly or more frequent follow-up examinations to check for the development of malignant tumors. Individuals with CCS along with their close family members should be offered access to in depth genetic counselling.[2] Research is needed to find the best treatments, including drugs, for these tumors.[2]
Signs and symptoms
Individuals with CCS generally have a family history of this disease and present with multiple (sometimes more than 100) benign hair follicle tumors most often on their head and torso.[7] The tumors usually begin at puberty and progressively accumulate throughout adulthood[2] although they have occurred in children as young as 1 year of age.[4] In severe cases, the tumors are highly disfiguring, extend throughout most of the scalp, face, and/or other hair-bearing sites such as the pubic area,[2] and uncommonly are malignant and may metastasize to non-cutaneous sites.[8] Milia, i.e. inconspicuous small cutaneous white spots caused by clogged eccrine sweat glands, may also occur in CCS and sometimes are the only indication that an individual has CCS; these individuals may develop tumors later in life and are at risk of having children with the full-blown disease.[2] CCS presentations are related to the microscopic histopathologies (see next section) of their tumors. i.e. their tumors may resemble non-familial cylindromas, spiradenomas, and/or trichoepitheliomas. Cylindroma-like CCS tumors appear as smooth nodular masses that are typically pink in color, may be translucent, may have superficial blood vessels, grow progressively over many years, and can reach several cm (i.e. centimeters) in size. Two or more of these tumors may merge to form far larger sized tumors. Spiradenoma-like CCS tumors also appear as nodular tumors but these may be blue/black in color, painful, rapidly growing and, like cylindromas, may merge to form large-size tumors. Features of both cylindroma-like and spiradenoma-like tumors (i.e. spiradenocylindromas) can occur in a single tumor. Trichoepithelioma-like CCS tumors appear as skin-coloured, small, papules usually located in the skin around the nose, nasolabial fold, and/or forehead. In individuals of European ancestry, the papules are usually 0.3-0.4 cm in size but in individuals of African, Indian, and Chinese ancestry may be larger.[2]
The MFT1 form of CCS typically presents with multiple trichoepithelioma-like tumors, the BSS form presents with cylindroma-, spiradenoma-, and/or trichoepithelioma-like tumors or papules, and the FC form typically presents with multiple cylindroma-like papules.[4] Uncommonly, individuals with CCS develop malignant tumors[9] that occur within or near to their benign tumors; they are primarily malignant counterparts to the cylindromas, spiradenomas, trichoepitheliomas and are termed cylindrocarcinomas, spiradenocarcinomas,[10] and trichoblastic carcinomas,[11][12] respectively. Some of these tumors resemble basal-cell carcinomas[13][14] or adenocarcinomas.[8] These malignant tumors may metastasize to non-cutaneous tissues such as the salivary glands (i.e.basal cell adenocarcinomas of a salivary gland[6]), liver, lungs, and/or bones.[2] Malignant CCS tumors occur more often in older individuals and tend to be larger (i.e. ranging up to 17.5 cm in size[7]) than their benign counterparts.[15]
Non-familial cylindromas,[16] spiradenomas,[7][13][17] spiradenocylindromas[7] and their malignant counterparts[7] present as a single isolated tumor or, less commonly, multiple tumors. These tumors are sporadic (i.e. not inherited) and may or may not consist of cells bearing a CYLD gene mutation. If present, the mutated CYLD gene's location is restricted to the tumor cells. These sporadic tumors are neither manifestations of nor diagnosed as CCS.[10]
Genetics
CCS is an autosomal dominant disorder inherited from a parent carrying an inactivating mutation in one of his or her two CYLD genes.[2] The mutation apparently causes hair follicle stem cells to differentiate into the cells which form cylindroma-, spiradenoma-, trichoepithelioma-, and/or spiradenocylindroma-like lesions.[8] Penetrance (i.e. the percentage of individuals with CCS that exhibit symptoms of the disease) has ranged from 44% to 100% (overall average, 72%) in different studies.[2] However, these studies usually assayed blood leukocyte samples for expression of the mRNA encoded by the CYLD gene to infer its presence. Recent studies find many cases of CCS involve mutations that go undetected by this method because they are: 1) mosaic mutations which distribute CYLD to blood leukocytes but little or none to hair follicles; 2) deep intronic mutations (i.e. mutations that occur deep within a CYLD gene's introns[18]) to result in the formation of mRNAs that do not code for an active protein but are detected as "normal" CYLD mRNA;[2] and 3) copy number variation mutations that result in the development of CYLD mRNA levels that vary widely in and within different tissues and in blood leukocytes samples may be high enough to give positive results but in hair follicles are too low to prevent tumor formation.[2][9] Genetic Tests which analyzed skin tumors rather than blood leukocytes, analyzed two or more skin tumors or blood leucocyte samples, and/or used sensitive next-generation sequencing of the CYLD gene have found evidence for inactivating CYLD gene mutations in cases that otherwise would be regarded as CYLD gene positive and therefore not CCS.[2][19]
Recent studies of 14 individuals with CCS indicate that their tumor cells have lost expression of both CYLD genes. This double gene mutation in CCS tumor cells was found in all benign and some malignant CCS tumors suggesting that it may be a consistent feature in CCS.[2][8] In this view, individuals with CCS carry an inherited, germline mutation in one CYLD gene that is widely distributed to the cells throughout the body plus am uninherited, acquired mutation in the second CYLD gene (see loss of heterozygosity) that is restricted to their CCS tumor cells. The lose of both CYLD genes in hair follicle stem cells may be required for the development of CCS tumors.[5]
CYLD gene
The CYLD gene is located in band 12.1 on the long (or "q") arm of chromosome 16.[4] It is classified as a tumor suppressor gene, i.e. a gene that regulates cell growth and when inactivated by a mutation leads to uncontrolled cell growth and the formation of tumors.[20] Inactivating CYLD gene mutations occur in T-Cell Acute Lymphoblastic Leukemia,[20] multiple myeloma, hepatocellular carcinoma, neuroblastoma, pancreatic cancer,[8] uterine cancer, stomach cancer, colon cancer, lung cancer, and human papillomavirus-associated cancers.[9] In a study of 95 mutations in CCS, 48% were frameshift, 27% were nonsense, 12% were missense, and 11% were splice site mutations.[5] Uncommonly, individuals with CCS have deep intronic mutations (i.e. mutations occurring in an intron that is more than 100 base pairs away from an exon–intron boundary)[2] or large scale mutations that delete most or all of the CYLD gene.[21] All of these mutations are inactivating mutations.[2][4][5][21]
The encoded product of the CYLD gene, CYLD lysine 63 deubiquitinase protein (CYLD protein), is a deubiquitinating enzyme, i.e. a protease that removes ubiquitin from certain proteins and thereby regulates these proteins' activities. CYLD protein removes ubiquitin from proteins involved in regulating the NF-κB, Wnt, notch, TGF-β,[2] and JNK[9] cell signaling pathways; these pathways normally act to regulate hair formation, cell growth, cell survival, inflammatory responses, and/or tumor development. The loss of CYLD protein's regulation of NF-κB signaling may be a critical contributor to the development of CCS tumors.[2][9] A wide range of stimuli cause cells to ubiquinate proteins that promote the movement of NF-κB from its inactive location in the cell's cytosol to the cell's nucleus. NF-κB is a transcription factor which when located in the nucleus stimulates the expression of various genes that in turn promote cell death by apoptosis and necroptosis[22] and stimulate cell growth.[23] CYLD protein releases ubiquitins from and thereby disables the proteins (e.g. IKBKG, also termed NEMO protien ) which promote cytosolic NF-κB movement to the nucleus. It is proposed that CCS tumor cells lack this restraint on NF-κB movements and therefore have abnormally prolonged survivals, high growth rates, and perhaps other features which contribute to tumor formation.[2][9]
Diagnosis
The diagnosis of CCS is strongly suggested by its tumors' presentations and microscopic histopathologies and the tumor bearers' family histories of CCS. Genetic testing to detect inactivating CYLD gene mutations is important for confirmation of the diagnosis.[2] Malignant CCS tumors are diagnosed based on the individuals personal and family history of CCS, the presence of metastases, and the microscopic histopathologicies of the malignant and metastatic tumors. Metastatic tumors may be directly tested for CYLD mutations to confirm the diagnosis.[10][14] To distinguish CCS from sporadic cases of cylindromas, spiradenomas, trichoepitheliomas, or their malignant counterparts, testing for the CYLD gene should be performed in individuals who have either: 1) two or more cylindromas, spiradenomas or trichoepitheliomas with at least one tumor being histologically confirmed or 2) a single (ideally histologically confirmed) cylindroma, spiradenoma or trichoepithelioma plus a family history of CCS confirmed by either genetic or histopathological evaluations.[2] In individuals suspected of having CCS but showing the presence of a normal CLYD gene based on the genetic testing of blood leukocytess, genetic testing of tumor tissue can confirm the diagnosis of CCS.[2][9] These methods differentiate CCS not only from sporadic cylindromas, spiradenomas, trichoepitheliomas, and their malignant counterparts but also from various skin tumor disorders with which they may be confused such as the Birt–Hogg–Dubé syndrome, tuberous sclerosis, neurofibromatosis, Cowden syndrome, Marie-Unna hypotrichosis, and multiple scalp trichilemmal cysts.[2]
Histopathology
As determined by the microscopic histopathological appearance of their hematoxylin and eosin-stained samples, CCS tumor tissues resemble sporadic cylindromas, spiradenomas, or trichoepitheliomas. Cylindroma-like CCS tumors are non-encapsulated nodular lesions that extend into the dermis, consist of basal cells (i.e. small, round cells similar to those seen in the lowest layer of the skin's epidermis), and are arranged in ("jigsaw-like"[10]) cylindrical patterns separated by thickened basement membranes. Spideradenoma-like CCS tumors consist of a relatively disorganized and dense array of proliferating basophilic cells (i.e. cells appearing blue because of their abnormally large uptake of the hematoxylin stain). Lymphocytes commonly populate these tumors' tissues. Some CCS tumors (i.e. spiradenocylindromas) merge the histopathological features of cylindromas and spiradenomas.[2] Trichoepithelioma-like CCS papules are composed of islands and cords of uniform basaloid cells (i.e. cuboid-shaped cells resembling skin germinative cells) in a fibrous stroma, epithelial structures resembling hair papillae or incompletely-formed hair follicles, small keratocysts (i.e. skin cysts) lined by stratified squamous epithelium, and foci of calcification.[14] The basiloid cells typically express epithelial cell adhesion molecule as detected by immunohistochemical analyses using the BerEp4 antibody. The tumor tissues of trichoepithelioma-like CCS tumors may also contain Merkel cells that express the CK20 protein.[10] Cylindrocarcinoma and spiradenocarcinoma tumors, unlike their benign counterparts, consist of low-grade or high-grade, atypical appearing cells that are rapidly proliferating (as defined, for example, by the cells showing high levels of the Ki67 protein marker of cell proliferation), may not express the Myb protein, and, with respect to spiradenocarcinoma tumors, usually do not have lymphocytes.[10] Trichoblastic carcinomas differ form their benign counterparts by consisting of hypercellular, fibrous, non-myxoid stroma, often resemble and may be diagnosed as basal cell carcinomas, and in high-grade tumors have extensive areas of necrosis (i.e. dead tissue); the cells in these tumors are rapidly proliferating.[11][12]
Treatment
CCS is an inherited, life-long, familial disease which currently is incurable.[4] All members of CCS families should be tested for the CYLD gene in order to identify those that do as well as those that do not carry an inactivating mutation in this gene. Testing for this gene is particularly important in cases with minimal evidence of the disease (e.g. Milia, see Presentation section) and may need to test tumor rather than blood leukocyte samples in individuals with mosaic, deep intronic, or copy number variation mutations (see Genetic section). All individuals who are found to carry the inactivated familial CYLD gene mutation should receive genetic counselling which may include: information on limitations in the ability to predict the severity of CCS between individuals, families, and generations; family planning; prenatal testing; preimplantation genetic diagnosis; and the need for follow-up evaluations. Individuals with CCS should get full-skin and possibly other organ (e.g. salivary gland) examinations performed yearly in routine cases, every 2–3 months in cases where their tumors show rapid growth or the formation of numaerous new skin tumors, and immediately in cases where their tumors develop signs of malignancy (e.g. tumor ulceration, very rapid growth, pain, intermittent bleeding, color changes, or tethering to an underlying bone or in cases that develop sudden onset of organ dysfunction such as breathlessness which may reflect lung metastases).[2]
Benign
The treatments for benign CCS tumors are directed at removing tumors that cause unacceptable symptoms (e.g. pain, ulceration, bleeding, disfigurement, sexual dysfunction, or occlusion of a passageway such as an ear canal).[2][24] Methods for these removals include surgical excision, electrodessication with curettage, dermabrasion, cryotherapy, or carbon dioxide, argon, or Er:YAG laser surgery.[4][14] Where indicated by cosmetic or functional considerations, these treatments are followed by plastic and/or reconstructive surgeries such as Flap surgery (i.e. normal tissue with its blood supply intact is lifted from a donor site and moved to the surgical site).[14] These treatments are often used repeatedly in individuals with numerous tumors, recurrent tumors, or newly formed tumors;[5] the treatments may also be extensive with, for example, up to 1 in 4 individuals with CCS requiring complete surgical removal of their scalps.[25]
Malignant
Primary CCS tumors that are known or suspected to be malignant and accessible metastatic tumors are currently treated with wide local excisions often followed by adjuvant radiotherapy. Wide local excision is used in order to remove all malignant cells while adjuvant radiotherapy is used to kill any malignant cells that remain behind after surgery.[7][13][14] Malignant CCS tumors uncommonly metastasize to distant, non-cutaneous sites. Histopathologically defined low-grade tumors metastasize less often than their high-grade counterparts.[10] Since past studies on the treatment of malignant CCS tumors often included, but did not clearly distinguish between, sporadic and familial cylindrocarcinoma, spiradenocarcinoma, and trichoblastic carcinoma tumors and since malignant CCS tumors are extremely rare, there are no clear data on the percentage of malignant CCS tumors that recur, metastasize, and become lethal. Clearly, however, CCS malignant tumors can produce each of these undesirable results.[7] The best treatment regimens for CCS malignant tumors are unclear and require further study.[5]
Medications
Trial studies have tested the therapeutic effects of drugs that inhibit the NF-κB pathway on CCS tumors. These drugs include topical (i.e. applied directly to the tumors) aspirin[6] or other salicylic acid-like drugs,.[5] In another drug trial study, an inhibitor of Trk receptors, topical Pegcantratinib, was similarly tested.[5][25] None of these treatments proved to have appreciable anti-tumor actions beyond occasional short-term effects.[5][6][25] One individual with CCS tumors was treat with Vismodegib, an inhibitor of the smoothened receptor and thereby the Hedgehog signaling pathway[26] and a second individual was treated with oral aspirin plus Adalimumab (a monoclonal antibody that neutralizes tumor necrosis factor alpha).[27] Both of these individuals showed slowly developing, modest improvements in there skin tumors although the individual treated with aspirin plus adalimumab showed no improvements in his multiple metastatic tumors. Further studies are needed to determine if these or any other drugs are effective in treating CCS tumors.[2]
See also
References
- ↑ Ransac S, Deveer AM, Rivière C, Slotboom AJ, Gancet C, Verger R, De Haas GH (January 1992). "Competitive inhibition of lipolytic enzymes. V. A monolayer study using enantiomeric acylamino analogues of phospholipids as potent competitive inhibitors of porcine pancreatic phospholipase A2". Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism. 1123 (1): 92–100. doi:10.1016/0005-2760(92)90175-u. PMID 1730050.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 Nagy N, Dubois A, Szell M, Rajan N (2021). "Genetic Testing in CYLD Cutaneous Syndrome: An Update". The Application of Clinical Genetics. 14: 427–444. doi:10.2147/TACG.S288274. PMC 8566010. PMID 34744449.
- ↑ Zaballos P, Gómez-Martín I, Martin JM, Bañuls J (October 2018). "Dermoscopy of Adnexal Tumors". Dermatologic Clinics. 36 (4): 397–412. doi:10.1016/j.det.2018.05.007. PMID 30201149. S2CID 52185272.
- 1 2 3 4 5 6 7 8 Arruda AP, Cardoso-Dos-Santos AC, Mariath LM, Feira MF, Kowalski TW, Bezerra KR, da Silva LA, Ribeiro EM, Schuler-Faccini L (July 2020). "A large family with CYLD cutaneous syndrome: medical genetics at the community level". Journal of Community Genetics. 11 (3): 279–284. doi:10.1007/s12687-019-00447-2. PMC 7295879. PMID 31792733.
- 1 2 3 4 5 6 7 8 9 Verhoeft KR, Ngan HL, Lui VW (2016). "The cylindromatosis (CYLD) gene and head and neck tumorigenesis". Cancers of the Head & Neck. 1: 10. doi:10.1186/s41199-016-0012-y. PMC 6460526. PMID 31093340.
- 1 2 3 4 Rajan N, Langtry JA, Ashworth A, Roberts C, Chapman P, Burn J, Trainer AH (November 2009). "Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction". Archives of Dermatology. 145 (11): 1277–84. doi:10.1001/archdermatol.2009.262. PMC 2935681. PMID 19917957.
- 1 2 3 4 5 6 7 Kazakov DV, Zelger B, Rütten A, Vazmitel M, Spagnolo DV, Kacerovska D, Vanecek T, Grossmann P, Sima R, Grayson W, Calonje E, Koren J, Mukensnabl P, Danis D, Michal M (May 2009). "Morphologic diversity of malignant neoplasms arising in preexisting spiradenoma, cylindroma, and spiradenocylindroma based on the study of 24 cases, sporadic or occurring in the setting of Brooke-Spiegler syndrome". The American Journal of Surgical Pathology. 33 (5): 705–19. doi:10.1097/PAS.0b013e3181966762. PMID 19194280. S2CID 9858971.
- 1 2 3 4 5 Davies HR, Hodgson K, Schwalbe E, Coxhead J, Sinclair N, Zou X, Cockell S, Husain A, Nik-Zainal S, Rajan N (October 2019). "Epigenetic modifiers DNMT3A and BCOR are recurrently mutated in CYLD cutaneous syndrome". Nature Communications. 10 (1): 4717. Bibcode:2019NatCo..10.4717D. doi:10.1038/s41467-019-12746-w. PMC 6797807. PMID 31624251.
- 1 2 3 4 5 6 7 Cui Z, Kang H, Grandis JR, Johnson DE (January 2021). "CYLD Alterations in the Tumorigenesis and Progression of Human Papillomavirus-Associated Head and Neck Cancers". Molecular Cancer Research : MCR. 19 (1): 14–24. doi:10.1158/1541-7786.MCR-20-0565. PMC 7840145. PMID 32883697.
- 1 2 3 4 5 6 7 Plotzke JM, Adams DJ, Harms PW (January 2022). "Molecular pathology of skin adnexal tumours". Histopathology. 80 (1): 166–183. doi:10.1111/his.14441. PMID 34197659. S2CID 235714739.
- 1 2 Boettler MA, Shahwan KT, Abidi NY, Carr DR (May 2021). "Trichoblastic carcinoma: a comprehensive review of the literature". Archives of Dermatological Research. doi:10.1007/s00403-021-02241-y. PMID 33993349. S2CID 234598207.
- 1 2 Janeczek M, Lehrer M, Pockaj B, DiCaudo D, Ochoa S (October 2021). "Metastatic trichoblastic carcinoma in the setting of trichoblastomatosis and multiple facial trichoepitheliomas". JAAD Case Reports. 16: 127–129. doi:10.1016/j.jdcr.2021.08.024. PMC 8455310. PMID 34584923.
- 1 2 3 Teli B, Thrishuli PB, Santhosh R, Amar DN, Rajpurohit S (2015). "Giant solitary trichoepithelioma". South Asian Journal of Cancer. 4 (1): 41–4. doi:10.4103/2278-330X.149951. PMC 4382785. PMID 25839021.
- 1 2 3 4 5 6 Kallam AR, Satyanarayana MA, Aryasomayajula S, Krishna BA (March 2016). "Basal Cell Carcinoma Developing from Trichoepithelioma: Review of Three Cases". Journal of Clinical and Diagnostic Research : JCDR. 10 (3): PD17–9. doi:10.7860/JCDR/2016/15432.7464. PMC 4843321. PMID 27134936.
- ↑ Srikantharajah T, Skovby F, Behrendt N, Jemec GB, Saunte DM (September 2020). "Can we clinically identify patients at risk of malignant transformation of skin tumors in Brooke-Spiegler syndrome?". Acta Dermatovenerologica Alpina, Pannonica, et Adriatica. 29 (3): 133–140. doi:10.15570/actaapa.2020.28. PMID 32975300. S2CID 221912295.
- ↑ Chauhan DS, Guruprasad Y (January 2012). "Dermal cylindroma of the scalp". National Journal of Maxillofacial Surgery. 3 (1): 59–61. doi:10.4103/0975-5950.102163. PMC 3513812. PMID 23251061.
- ↑ Vy M, Mehrzad M, Konia T, Burrall B (May 2021). "An unusual case of multiple grouped non-familial trichoepitheliomas". Dermatology Online Journal. 27 (5). doi:10.5070/D327553615. PMID 34118815. S2CID 235426045.
- ↑ Vaz-Drago R, Custódio N, Carmo-Fonseca M (September 2017). "Deep intronic mutations and human disease". Human Genetics. 136 (9): 1093–1111. doi:10.1007/s00439-017-1809-4. PMID 28497172. S2CID 3725196.
- ↑ Arefi M, Wilson V, Muthiah S, Zwolinski S, Bajwa D, Brennan P, Blasdale K, Bourn D, Burn J, Santibanez-Koref M, Rajan N (December 2019). "Diverse presentations of cutaneous mosaicism occur in CYLD cutaneous syndrome and may result in parent-to-child transmission". Journal of the American Academy of Dermatology. 81 (6): 1300–1307. doi:10.1016/j.jaad.2019.05.021. PMC 6878220. PMID 31085270.
- 1 2 Lei H, Wang J, Hu J, Zhu Q, Wu Y (August 2021). "Deubiquitinases in hematological malignancies". Biomarker Research. 9 (1): 66. doi:10.1186/s40364-021-00320-w. PMC 8401176. PMID 34454635.
- 1 2 Zhu R, Xu J, Shen J, Li W, Tan F, Li C, Wei Z, Liu Y, Bai Y (October 2020). "A novel large deletion of the CYLD gene causes CYLD cutaneous syndrome in a Chinese family". Molecular Genetics & Genomic Medicine. 8 (10): e1441. doi:10.1002/mgg3.1441. PMC 7549610. PMID 32783365.
- ↑ Lork M, Verhelst K, Beyaert R (July 2017). "CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different". Cell Death and Differentiation. 24 (7): 1172–1183. doi:10.1038/cdd.2017.46. PMC 5520167. PMID 28362430.
- ↑ Vlahopoulos SA (August 2017). "Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode". Cancer Biology & Medicine. 14 (3): 254–270. doi:10.20892/j.issn.2095-3941.2017.0029. PMC 5570602. PMID 28884042.
- ↑ Rajan N, Trainer AH, Burn J, Langtry JA (May 2009). "Familial cylindromatosis and brooke-spiegler syndrome: a review of current therapeutic approaches and the surgical challenges posed by two affected families". Dermatologic Surgery : Official Publication for American Society for Dermatologic Surgery [Et Al.] 35 (5): 845–52. doi:10.1111/j.1524-4725.2009.01142.x. PMID 19397670. S2CID 10361680.
- 1 2 3 Danilenko M, Stamp E, Stocken DD, Husain A, Zangarini M, Cranston A, Stones R, Sinclair N, Hodgson K, Bowett SA, Roblin D, Traversa S, Plummer R, Veal G, Langtry JA, Ashworth A, Burn J, Rajan N (August 2018). "Targeting Tropomyosin Receptor Kinase in Cutaneous CYLD Defective Tumors With Pegcantratinib: The TRAC Randomized Clinical Trial". JAMA Dermatology. 154 (8): 913–921. doi:10.1001/jamadermatol.2018.1610. PMC 6128505. PMID 29955768.
- ↑ Baur V, Papadopoulos T, Kazakov DV, Agaimy A, Hartmann A, Isbary G, Wirtz RM, Schultz ES (August 2018). "A case of multiple familial trichoepitheliomas responding to treatment with the Hedgehog signaling pathway inhibitor vismodegib". Virchows Archiv : An International Journal of Pathology. 473 (2): 241–246. doi:10.1007/s00428-018-2397-y. PMID 29934657. S2CID 49383828.
- ↑ Fisher GH, Geronemus RG (June 2006). "Treatment of multiple familial trichoepitheliomas with a combination of aspirin and a neutralizing antibody to tumor necrosis factor alpha: A case report and hypothesis of mechanism". Archives of Dermatology. 142 (6): 782–3. doi:10.1001/archderm.142.6.782. PMID 16785388.