Discovery and development of neuraminidase inhibitors
Neuraminidase inhibitors inhibit enzymatic activity of the enzyme neuraminidase (sialidase). These type of inhibitors have been introduced as anti-influenza drugs as they prevent the virus from exiting infected cells and thus stop further spreading of the virus. Neuraminidase inhibitors for human neuraminidase (hNEU) have the potential to be useful drugs as the enzyme plays a role in several signaling pathways in cells and is implicated in diseases such as diabetes and cancer.[1]
History
The first neuraminidase Inhibitors (NAIs) were synthesized in the 1960s by Edmond et al.,[2] through an attempt to understand the catalytic mechanism of the neuraminidase enzyme. They discovered that N-substituted oxamic acids had enzyme inhibitory properties. Then it was found that the synthetic compound 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (Neu5Ac2en or DANA) which is an analogue of N-acetylneuraminic acid (Neu5Ac), inhibits the release of virus progeny in tissue culture but no antiviral activity in animals was detected.[3][4] In the early 1990s, the determination of biological crystal structure of influenza virus surface protein led to the discovery of the active site and provided the opportunities to discover and design new and specific inhibitors.
Influenza virus
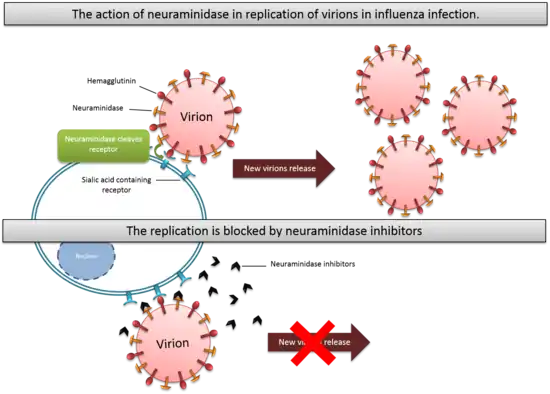
The Influenza virus is an RNA virus that is divided into three serological types: A, B and C. Hemagglutinin (HA) and neuraminidase (NA) are two important glycoproteins on influenza virus membranes. The hemagglutinin is a sialic acid receptor-binding molecule and mediates entry of the virus into the host cell, while neuraminidase cleaves cellular-receptor sialic acid to form new particles. Neuraminidase is an exoglycosidase that destroys the hemagglutinin receptor by cleaving the α(2,6)- or α(2,3)-ketosidic linkage that exists between a terminal sialic acid and a sugar residue of the Neu5Ac containing receptor on the surface of host cells.[5] This helps the spread of the infection by preventing self-aggregation of new viruses at the cell surface and possible immobilisation in the mucin by hemagglutinin (HA) during virus replication. The virus will then be released from the host cells and will subsequently infect other cells.[6] Neuraminidase also helps the invasion of the virus in the upper respiratory tract, possibly by cleaving sialic acid molecules on mucin of epithelial cells. Neuraminidase is found in influenza viruses of types A and B.[7] Neuraminidase has roles in the infection, replication and delivery of Influenza virus A and B. Type C Influenza virus expresses the enzyme esterase instead of neuraminidase.[8]
The substrate
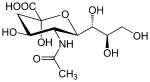
N-acetylneuraminic acid (Neu5Ac) is one of the two most common sialic acid in mammals.[9] It is a monosaccharide with a backbone of 9 carbons. It is usually attached to glycoproteins or gangliosides on a terminal end via α(2,3), α(2,6), and α(2,8) linkage.[10] Neuraminidase is an enzyme which hydrolyses that bond to produce a free neuraminic acid and a glycoprotein or a sugar chain. Influenza virus will bind via the hemagglutinin protein on these sialic acid attached glycoproteins on the cell membrane.[11]
Mechanism of action
Mechanism of enzyme catalysis
The mechanism of NA has been shown to proceed with the retention of configuration which means it preserves the absolute configuration on the atom in the stereocenter.[12] There are four steps of catalytic pathways. In the first step, the binding step, the carboxylate group changes from the axial position into the pseudo-equatorial position. The second step is the proton donation from water molecule and formation of the endocyclic sialosyl cation transition-state intermediate. Step three involves nucleophilic attack of tyrosine on the sialosyl cation. The fourth step is the formation and release of Neu5Ac. A similar mechanism has been proposed by Janakiraman et al.[13] where the double bond of Neu5Ac2en forces the pyranose of sugar ring into a planar structure were resembled the transition-state structure.
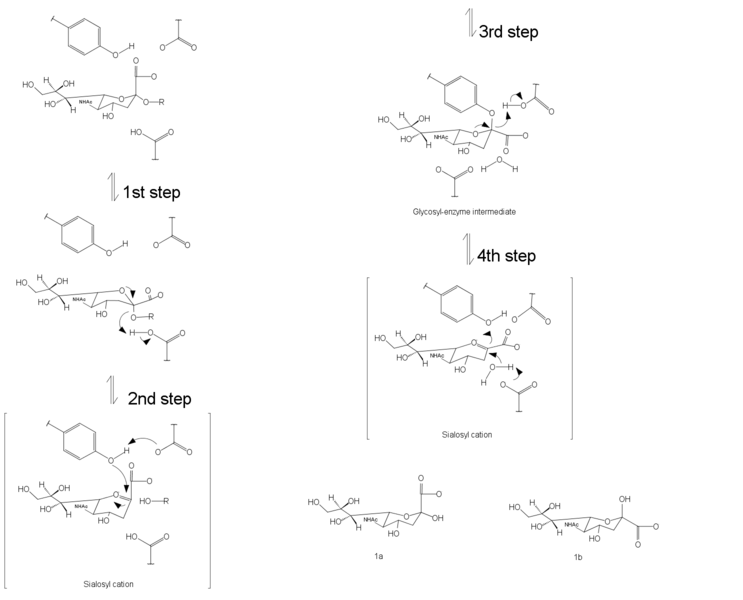
Mechanism of inhibition
There are two types of neuraminidase inhibitors commonly available for treatment and prophylaxis of influenza infections: Zanamivir and Oseltamivir. They interfere with the release of progeny virions from infected host cells, prevent infection process of new host cells and stop spread of infection in respiratory tract by mimicking natural substrate and fitting into active site of neuraminidase enzyme. They interrupt the detachment of progeny virions. Viral replication rate is then reduced and that allows human immune system to destroy the remaining viruses.[14]
Development
Viral neuraminidase
.png.webp)
Influenza virus neuraminidase (vNEU) consists of 4 co-planar roughly spherical subunits predominantly made of β-sheets, characterized as a 6-fold β-propeller and a hydrophobic region embedded in the virus’ membrane.[15] The active site is located near the middle of the pseudo-symmetric sphere. Influenza virus neuraminidase only cleaves terminal Neu5Ac residues. X-ray crystallography has shown a distorted half-chair arrangement of the Neu5Ac substrate in the active site. This distorted structure forms a sialosyl cation after the release of the aglycon and is then trapped in the active site by a nucleophilic attack of the tyrosine residue. The orientation of the substrate in the active site is facilitated mainly by three strain-preserved Arginine residues binding the C1 acid group with salt bridges. Furthermore, the active site consists of eight other highly conserved amino acid-residues that make direct contact to the substrate or its derivatives. Including a glutamic acid residue binding the C7 and C9 alcohol groups on the glycerol side-chain (at C6) with hydrogen bonds and several hydrophobic residues correlating with the methyl group on the C5 N-acetyl and the hydrophobic backbone of the glycerol.[16]
Human Neuraminidase
Human neuraminidase (hNEU) shares many similar features with vNEU. The human genome has four different neuraminidase enzymes (NEU1, NEU2, NEU3, NEU4) and only one of them (NEU2) is not membrane-associated or in a membrane-complex and has been studied with X-ray crystallography.[1][17] The three arginine residues that bind the C1 acid-group with salt bridges in vNEU are also present in hNEU. Active site topology and interactions with the substrate are very similar with the exception of the glycerol side chain which offers some strategic options in designing inhibitors targeting either vNEU or hNEU. In hNEU the glycerol hydroxyl-groups are bound via several tyrosine residues but in vNEU the main interaction is with a glutamic acid residue. These overall similarities have called for concerns over potential side effects from drugs targeting vNEU. Nevertheless, most of the well studied vNEU inhibitors have shown very little affinity for hNEU except for the influenza drug Zanamivir which is an effective inhibitor for hNEU2.[1][16]
Viral neuraminidase inhibitors
2-deoxy-2,3-didehydro-N-acetylneuraminic acid (Neu5Ac2en) is a pan-selective inhibitor for neuraminidase. Neu5Ac2en is a dehydrogenated Neu5Ac and can be synthesized by the hNEU enzyme if Neu5Ac is in high enough concentration. Neu5Ac is also a mild inhibitor for the enzyme but as Neu5Ac2en is a transition-state analogue it is a much better inhibitor.[1]
Zanamivir
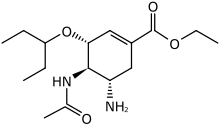
In the beginning the X-ray crystallography did not have a very good resolution so the initial focus was on substrate derived inhibitors instead of structurally based.[18] The Neu5Ac-derived 2-deoxy-α-D-N-acetylneuraminic acid (2-deoxy- α-Neu5Ac) was the first template used and also the first inhibitor tried in vivo in a mouse model of an influenza infection. The unsubstituted template showed minor effect. Another template Neu5Ac2en (DANA) was tried under same conditions and showed good in vivo effect. With new crystal structure images of the enzyme and Neu5Ac complex emerging and Neu5Ac2en confirmed as an in vivo inhibitor, the focus was on making structure based DANA derivatives. With better X-ray crystal structure a number of important residues in the active site were identified, specifically C4 hydroxyl group. Better effect was achieved by substituting the C4 hydroxyl group with a more basic group, for example an amino group. Further analysis showed that a larger group could be accommodated in the active site. 4-amino-4-deoxy-Neu5Ac2en and 4-deoxy-4-guanidino-Neu5Ac2en were synthesized and proved to be competitive inhibitors for viral neuraminidase and significantly inhibited both A and B influenza replication in vitro and in vivo. 4-deoxy-4-guanidino-Neu5Ac2en showed not only to be the better inhibitor but also showed considerable lower affinity for other isoforms of neuraminidase. For these reasons 4-deoxy-4-guanidino-Neu5Ac2en was selected as the main drug candidate under the name Zanamivir. High polar nature and rapid excretion contribute to the drugs low bioavailability and rapid elimination.[6][16][19]
Oseltamivir
Multiple new inhibitors based on non-carbohydrate templates have been synthesized. With focus on positioning the double bond in the inhibitor to more closely resemble the transition state of the substrate and replacing the glycerol side chain with a lipophilic group on the basis of the hydrophobic backbone of the glycerol interacting with the protein lead to the discovery of GS 4071. GS 4071 is cyclohexene based and has 3-pentyl ether, found to be optimal, instead of the glycerol as the side chain. The GS 4071 inhibitor is more lipophilic than the predecessor Zanamivir but does not have more bioavailability. Oseltamivir, the ethyl ester of GS 4071 was produced as a prodrug and is actively converted to the active drug in vivo.[6][16][19]
Peramivir
Peramivir is developed by structure-based drug design. After the influenza NA inhibitor activity of α/β-6-acetyl-amino-3,6-dideoxy -D-glycero-altro-2-nonulofuranosonic acid was reported by Yamamoto et al.,[20] the cyclopentane derivatives was designed with a guanidino group replacing C4-hydroxyl position of DANA in the active site, similar to Zanamivir. Babu et al.[18] found that the addition of n-butyl side chain makes the compound fit better to the hydrophobic region of the enzyme. However, the conformation of the n-butyl group was found to be different when bound to influenza virus A from its conformation when bound to influenza virus B. [21] Since the compound processed similar binding interaction with active site of NA to zanamivir and due to the mutation in some zanamivir-resistance strains, the position of guanidino group was altered and the n-butyl group was replaced in order to change its active site interaction.[18][22]
Structures of the Viral neuraminidase inhibitors in use
 |
 |
 |
 |
| Zanamivir | Oseltamivir | Peramivir | Laninamivir |
*Only Zanamivir and Oseltamivir are FDA approved. Peramivir is used in Japan and South Korea. Laninamivir is used in Japan only.[23]
Recent development and design of analogues of viral inhibitors
New NA inhibitor analogues were synthesized, based on Zanamivir, Oseltamivir and Peramivir, with rational structure-based drug design and can be categorized into four groups.
Analogues of Zanamivir
Zanamivir analogues are designed to improve the therapeutic use. Replacing the carboxylate group at the C1 to phosphonate group led the drug to be more potent with high affinity to form ionic interaction with the active site. Additionally, the click-chemistry reaction was used to synthesize the C4-triazole-modified zanamivir analogue that shows inhibitory activity close to zanamivir.[24] Laninamivir is designed by replacing C7 hydroxy moiety with small lipophilic group, -OCH3, which resulted in an excellent inhibitory activity. The C8 and C9 diol play an important role in the binding affinity with neuraminidase, prolonging the effect. The polymer scaffolds at the C7 position of zanamivir via an alkyl ether has gained more attention as this showed enhanced antiviral activity.[25]
 |
 |
| C4-trizole | 4-aminophosphono-DANA |
 |
 |
| Phosphono-zanamivir | Prodrug of Phosphono-zanamivir |
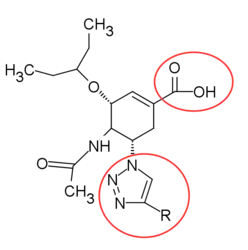
Analogues of Oseltamivir
The knowledge of transition state structure has been used to design oseltamivir analogues. For example, the triazole-containing carbocycles by Von Itzstein and Pinto group and the phosphonate analogue of oseltamivir has been reported to show stronger activity resulted from a pertinent binding mode of phosphonate with three Arginine residues in the active site.[26]
 |
 |
| Triazole-containing carbocycles analogue | Phosphonate analogue. R1, R2 = -H or -CH2CH3 |
,
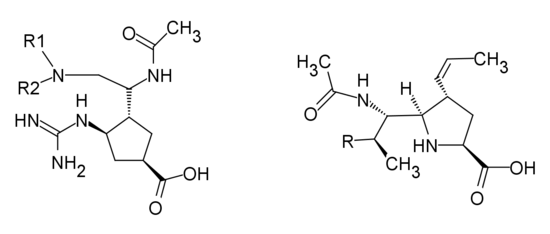
Analogues of Peramivir
Peramivir is the cyclopentane derivative designed with a guanidino group replacing the C-4 hydroxyl group of Neu5Ac2en in the active site, with negatively charged carboxylate group and a n-butyl side chain. 1-ethylpropylamide, diethylamide, dipropylamide and 4-morpholinylamide groups all showed excellent inhibitory activity. Changing the cyclopentane ring to a pyrrolidine ring showed high inhibitory activity as well.[27]
 |
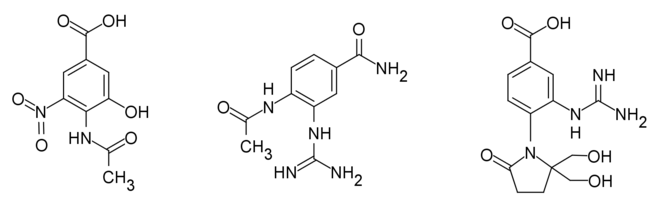
Analogues of Benzoic acid
The replacement of dihydropyran ring with a benzene ring increases the lipophilicity of a drug and makes the drug much more stable and more synthetically accessible. Based on the interaction of DANA and NA, the benzoic derivatives were synthesized. They have shown similar or better inhibitory activity compared to Neu5Ac.[26]
 |
Human neuraminidase inhibitors
Zanamivir and Oseltamivir have been tested as hNEU inhibitors. Only Zanamivir shows moderate inhibition activity for hNEU. Isoenzyme selective inhibitors could potentially be very important. At present there are limited studies for the hNEU substrate specificity. DANA is a pan-selective inhibitor for all hNEU isoenzymes, with a difference from 2 to 10 fold inhibition activity, most effective for hNEU3 and hNEU4. Several reports have tested DANA-derivatives as inhibitors for hNEU2 and hNEU3. Most derivatives showed reduced inhibition in vitro. However, N5-azidoacetate-C9-azido derivative of DANA showed improved inhibition for hNEU2 and hNEU3. These studies concluded that the active site in the enzymes could tolerate large modifications at C9 better than at N5.[1] Testing of C9 amido derivatives of DANA showed hNEU1-selective inhibitors. These compounds showed more activity then DANA and only minor activity for hNEU3 with around 25-fold selectivity for hNEU1. These studies show that the glycerol side-chain pocket in the active site can potentially be manipulated in the design of isoenzyme selective inhibitors for hNEU. A combination of C4 and C7 modified DANA derivatives has been reported with moderate selectivity for hNEU2 and hNEU3.[28] Finally C9-triazole derivatives of DANA containing an alkoxy group with a nanomolar activity against hNEU4 isoenzyme have been reported. With a 500-fold selectivity for hNEU4, this is the highest reported selectivity for a hNEU isoenzyme to date.[1]
Usage
There are 2 subgroups of NA inhibitors that have been approved by regulatory authorities in the US and Europe, Zanamivir and Oseltamivir. Both are for the treatment and prevention of influenza. Furthermore, Peramivir and Laninamivir have been approved by regulatory authorities in some parts of Asia.[23]
Laninamivir
Laninamivir is approved for the treatment of influenza under the tradename Inavir in Japan but it is still in clinical trial in the USA.[29] Laninamivir is a long acting inhaled drug given as a prodrug (laninamivir octanoate). Laninamivir is given as a single dose and remains active for at least 5 days and up to 7 days.[30][31]
Oseltamivir
Oseltamivir can be found under tradenames such as Agucort®, Antiflu, Fluvir, Fluhalt, GPO-A-Flu™, Omiflu, Rimivat, Virobin, Oseltamivir and Tamiflu®.[32] Oseltamivir is used for patients 1 year and older. It is given as one dose, twice a day for the treatment of influenza. In the prevention of influenza, oseltamivir is given as one dose, once a day for at least 10 days after contacting with an infected person and up to six months (during influenza epidemic). The most common side effects of Oseltamivir are headache and nausea (in adults) and vomiting, cough and nasal congestion (in children).[33]
Peramivir
Peramivir is approved for the treatment of influenza under the tradenames Rapiacta® in Japan and Peramiflu in South Korea.[34] In the US and elsewhere, peramivir is undergoing the late-phase clinical trial. Peramivir is used as intravenous and was used in the emergency treatment of 2009 H1N1 in select patients.[30][35]
Zanamivir
Zanamivir can be found under tradenames such as Relenza™, Verenza and Z-Flu DPI.[36] Zanamivir is used for patients 5 years and older. It is given as one 10 mg dose, twice a day for the treatment of influenza. In the prevention of influenza, zanamivir is given as one 10 mg dose, once a day for 10 days after contacting with an infected person or up the 28 days (during influenza epidemic). The most common side effect of Zanamivir is reported to be rash.[37]
Drug resistance
Currently, there are two classes of antiviral drugs approved for the treatment and prophylaxis of influenza infections. They are the adamantanes and NAIs. The adamantanes only work on influenza A so since 2010 WHO recommended the usage of NAIs for treatment and prophylaxis of influenza A and B infections.[38] In contrast to adamantanes, NAIs are less toxic and less prone to promote drug-resistant influenza. Moreover, they are effective against all neuraminidase subtypes and all strains of influenza. After the influenza pandemic in 2009, there has been great concern about viral resistance to NAIs.[30][38][39] Influenza viruses that have reduced sensitivity to NAIs often contain mutation that affect the shape of the NA catalytic site and therefore reduce the binding ability of the inhibitors. The catalytic site of the NA has eight functional residues ( R118, D151, R152, R224, E276, R292, R371, and Y406) surrounded by eleven framework residues (E119, R156, W178, S179, D198, I222, E227, H274, E277, N294, and E425).[38]
Resistance to Oseltamivir
Oseltamivir has a large hydrophobic side chain and the NA must undergo rearrangement to form a pocket for drug binding by rotating aminoacid E276 and bond with R224. Mutations like H274Y, R292K and N294S that affect this forming could reduce the inhibitor's efficiency.[38]
Resistance to Zanamivir
Resistance to zanamivir has been low for both seasonal and pandemic viruses compare to oseltamivir. Molecular structure of zanamivir has a guanidino group, this group interacts with the E119 residue in the active center pocket. Resistance to zanamivir can be because of mutations that effect binding affinity between the enzyme and the inhibitor. Mutation at the E119 residue has been shown to reduce the inhibitors efficiency in vitro.[38]
Resistance to Peramivir
Peramivir has a guanidino group similar to zanamivir and a hydrophobic group similar to oseltamivir. Mutations that effect the efficiency of oseltamivir and zanamivir can also effect peramivir efficiency. Resistances to peramivir have been seen at the mutation of H274Y residue in vitro. One of these resistances is associated with cross-resistance to peramivir and oseltamivir.[38] Peramivir is approved in Japan as Rapiacta and also available in South Korea as Peramiflu.[23]
Resistance to Laninamivir
No laninamivir resistance has been reported. However it is a concern that resistance to laninamivir is similar to that of zanamivir because of the similarity in binding properties with the NA protein.[38] Laninamivir octanoate (CS-8958), which is a prodrug of laninamivir (another inhaled NAI with long-acting properties), has also been approved in Japan and is commercially available under the name of Inavir (Daiichi Sankyo Company Ltd.[23]
See also
Neuraminidase
Neuraminidase inhibitors
Influenza virus
Adamantane
References
- Christopher W. Cairo. (2014) Inhibitors of the human neuraminidase enzymes. Med. Chem. Commun., 2014, 5, 1067.DOI: 10.1039/c4md00089g
- Edmond, J. D., Johnston, R. G., Kidd, D., Rylance, H. J. and Sommerville, R. G. (1966) The Inhibition Of Neuraminidase And Antiviral Action. Br. J. Pharmacol. Chemother., 1966, 27: 415–426. doi: 10.1111/j.1476-5381.1966.tb01673.x
- Kim, Choung U, Xiaowu Chen, and Dirk B Mendel. Neuraminidase inhibitors as anti-influenza virus agents. Antiviral chemistry & chemotherapy 10.4 (1999): 41-154
- von Itzstein, Mark. The war against influenza: discovery and development of sialidase inhibitors. Nature reviews Drug discovery 6.12 (2007): 967-974
- Air, Gillian M, and W Graeme Laver. The neuraminidase of influenza virus. Proteins: Structure, Function, and Bioinformatics 6.4 (1989): 341-356
- Varghese, J. N. (1999). Development of neuraminidase inhibitors as anti-influenza virus drugs. Drug Development Research, 46(3-4), 176-196
- Du, Juan, Timothy A Cross, and Huan-Xiang Zhou. Recent progress in structure-based anti-influenza drug design. Drug discovery today 17.19 (2012): 1111-1120
- Wagaman PC, Spence HA, and O’Callaghan RJ. 1989. Detection of Influenza C Virus by Using an In Situ Esterase Assay Journal of Clinical Microbiology. 1:832-836.
- Varki, N. M., & Varki, A. (2007). Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab Invest, 87(9), 851-857. doi: 10.1038/labinvest.3700656
- Varki, Ajit; Roland Schauer (2008). in Essentials of Glycobiology. Cold Spring Harbor Press. pp. Ch. 14
- Racaniello, Vincent. (2009). Influenza virus attachment to cells. Retrieved October 2014, from http://www.virology.ws/2009/05/04/influenza-virus-attachment-to-cells/
- Taylor, N. R., & von Itzstein, M. (1994). Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. Journal of medicinal chemistry, 37(5), 616-624
- Janakiraman, M. N., White, C. L., Laver, W. G., Air, G. M., & Luo, M. (1994). Structure of Influenza Virus Neuraminidase B/Lee/40 Complexed with Sialic Acid and a Dehydro Analog at 1.8-. ANG. Resolution: Implications for the Catalytic Mechanism. Biochemistry, 33(27), 8172-8179
- Moscona, A. (2005). Neuraminidase Inhibitors for Influenza. New England Journal of Medicine, 353(13), 1363-1373. doi: doi:10.1056/NEJMra050740
- Colman, P.M. (1994) Influenza virus neuraminidase: Structure, antibodies, and inhibitors.Protein Science. 3: 1687–1696. doi: 10.1002/pro.5560031007
- Mark von Itzstein (2007)The war against influenza: discovery and development of sialidase inhibitors. Nature publishing group. Volume 6. 967-974
- havas LMG, Tringali C, Fusi P, Venerando B, Tettamanti G, Kato R, Monti E, Wakatsuki S. Crystal structure of the human cytosolic sialidase Neu2 - Evidence for the dynamic nature of substrate recognition. J Biol Chem. 2005;280:469-475.
- Babu, Y. S. (and 13 others) 2000 BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective in£uenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 43, 3482^3486.
- Varghese, J. N.; Smith, P. W.; Sollis, S. L.; Blick, T. J.; Sahasrabudhe, A.; McKimm-Breschkin, J. L.; Colman, P. M. (1998). "Drug design against a shifting target: A structural basis for resistance to inhibitors in a variant of influenza virus neuraminidase". Structure. 6 (6): 735–746. doi:10.1016/S0969-2126(98)00075-6. PMID 9655825.
- Yamamoto, T., Kumazawa, H., Inami, K., Teshima, T. & Shiba, T. 1992. Syntheses of sialic acid isomers with inhib- itory activity against neuraminidase. Tetrahedron Lett. 33, 5791^5794
- Smee, D. F., Huffman, J. H., Morrison, A. C., Barnard, D. L., & Sidwell, R. W. (2001). Cyclopentane neuraminidase inhibitors with potent in vitro anti-influenza virus activities. Antimicrob Agents Chemother, 45(3), 743-748. doi: 10.1128/AAC.45.3.743-748.2001
- Young, Diane, Cynthia Fowler, and Karen Bush. RWJ-270201 (BCX-1812): a novel neuraminidase inhibitor for influenza. Philos Trans R Soc Lond B Biol Sci 356.1416 (2001): 1905-1913
- Burnham, A. J., Baranovich, T., & Govorkova, E. A. (2013). Neuraminidase inhibitors for influenza B virus infection: efficacy and resistance. Antiviral Res, 100(2), 520-534. doi: 10.1016/j.antiviral.2013.08.023
- Abdel-Magid, Ahmed F, Cynthia A Maryanoff, and Steven J Mehrman. Synthesis of influenza neuraminidase inhibitors. Current Opinion in Drug Discovery & Development 4.6 (2001): 776-791.
- Russell, Rupert J et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443.7107 (2006): 45-49
- Feng, Enguang et al. Recent Advances in Neuraminidase Inhibitor Development as Anti‐influenza Drugs. ChemMedChem 7.9 (2012): 1527-1536
- Chand, Pooran et al. Systematic structure-based design and stereoselective synthesis of novel multisubstituted cyclopentane derivatives with potent antiinfluenza activity. Journal of medicinal chemistry 44.25 (2001): 4379-4392
- Albohy, A., Zhang, Y., Smutova, V., Pshezhetsky, A. V., & Cairo, C. W. (2013). Identification of Selective Nanomolar Inhibitors of the Human Neuraminidase, NEU4. ACS Medicinal Chemistry Letters, 4(6), 532–537. doi:10.1021/ml400080t
- Laninamivir octanoate (LANI) – Influenza.). Retrieved October 27th, 2014, from http://www.biotapharma.com/index.php/pipeline
- Hurt, A. C. (2014). The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol, 8C, 22-29. doi: 10.1016/j.coviro.2014.04.009
- Ikematsu, H., & Kawai, N. (2011). Laninamivir octanoate: a new long-acting neuraminidase inhibitor for the treatment of influenza. Expert Rev Anti Infect Ther, 9(10), 851-857. doi: 10.1586/eri.11.112
- J05AH02 - Oseltamivir. (2014). Retrieved November 2nd, 2014, from http://www.medicatione.com/?c=atc&s=j05ah02
- Agency, European Medicines. (2012). Tamiflu. Retrieved October 15th, from European Medicines Agency http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000402/WC500033101.pdf
- Peramivir.). Retrieved October 27th, 2014, from http://www.biocryst.com/peramivir Archived 2014-11-16 at the Wayback Machine
- Mancuso, C. E., Gabay, M. P., Steinke, L. M., & Vanosdol, S. J. (2010). Peramivir: an intravenous neuraminidase inhibitor for the treatment of 2009 H1N1 influenza. Ann Pharmacother, 44(7-8), 1240-1249. doi: 10.1345/aph.1P031
- J05AH01 - Zanamivir. (2014). Retrieved November 2nd, 2014, from http://www.medicatione.com/?c=atc&s=j05ah01
- Icelandic Medicines Agency. (2014). Samantek á eiginleikum lyfs Retrieved October 15th from Icelandic Medicines Agency "Archived copy" (PDF). Archived from the original (PDF) on 2014-12-25. Retrieved 2014-11-04.
{{cite web}}: CS1 maint: archived copy as title (link) - Samson, M., Pizzorno, A., Abed, Y., & Boivin, G. (2013). Influenza virus resistance to neuraminidase inhibitors. Antiviral Res, 98(2), 174-185. doi: 10.1016/j.antiviral.2013.03.014
- Dixit, R., Khandaker, G., Ilgoutz, S., Rashid, H., & Booy, R. (2013). Emergence of oseltamivir resistance: control and management of influenza before, during and after the pandemic. Infect Disord Drug Targets, 13(1), 34-45