Epigenetics
In biology, epigenetics is the study of stable phenotypic changes (known as marks) that do not involve alterations in the DNA sequence.[1] The Greek prefix epi- (ἐπι- "over, outside of, around") in epigenetics implies features that are "on top of" or "in addition to" the traditional genetic basis for inheritance.[2] Epigenetics most often involves changes that affect gene activity and expression, but the term can also be used to describe any heritable phenotypic change. Such effects on cellular and physiological phenotypic traits may result from external or environmental factors, or be part of normal development.
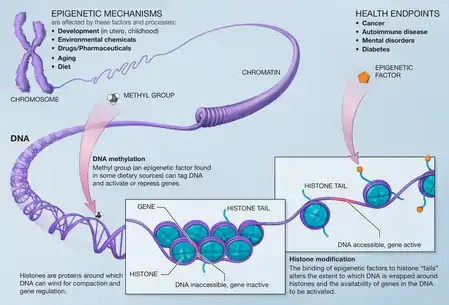
The term also refers to the changes themselves: functionally relevant changes to the genome that do not involve a change in the nucleotide sequence. Examples of mechanisms that produce such changes are DNA methylation and histone modification, each of which alters how genes are expressed without altering the underlying DNA sequence. Gene expression can be controlled through the action of repressor proteins that attach to silencer regions of the DNA. These epigenetic changes may last through cell divisions for the duration of the cell's life, and may also last for multiple generations, even though they do not involve changes in the underlying DNA sequence of the organism;[3] instead, non-genetic factors cause the organism's genes to behave (or "express themselves") differently.[4]
One example of an epigenetic change in eukaryotic biology is the process of cellular differentiation. During morphogenesis, totipotent stem cells become the various pluripotent cell lines of the embryo, which in turn become fully differentiated cells. In other words, as a single fertilized egg cell – the zygote – continues to divide, the resulting daughter cells change into all the different cell types in an organism, including neurons, muscle cells, epithelium, endothelium of blood vessels, etc., by activating some genes while inhibiting the expression of others.[5]
Definitions
The term epigenetics in its contemporary usage emerged in the 1990s, but for some years has been used with somewhat variable meanings.[6] A definition of the concept of epigenetic trait as a "stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence" was formulated at a Cold Spring Harbor meeting in 2008,[7] although alternate definitions that include non-heritable traits are still being used widely.[8]
The term epigenesis has a generic meaning of "extra growth" that has been used in English since the 17th century.[9]
Waddington's canalisation, 1940s
The hypothesis of epigenetic changes affecting the expression of chromosomes was put forth by the Russian biologist Nikolai Koltsov.[10] From the generic meaning, and the associated adjective epigenetic, British embryologist C. H. Waddington coined the term epigenetics in 1942 as pertaining to epigenesis, in parallel to Valentin Haecker's 'phenogenetics' (Phänogenetik).[11] Epigenesis in the context of the biology of that period referred to the differentiation of cells from their initial totipotent state during embryonic development.[12]
When Waddington coined the term, the physical nature of genes and their role in heredity was not known. He used it instead as a conceptual model of how genetic components might interact with their surroundings to produce a phenotype; he used the phrase "epigenetic landscape" as a metaphor for biological development. Waddington held that cell fates were established during development in a process he called canalisation much as a marble rolls down to the point of lowest local elevation.[13] Waddington suggested visualising increasing irreversibility of cell type differentiation as ridges rising between the valleys where the marbles (analogous to cells) are travelling.[14]
In recent times, Waddington's notion of the epigenetic landscape has been rigorously formalized in the context of the systems dynamics state approach to the study of cell-fate.[15][16] Cell-fate determination is predicted to exhibit certain dynamics, such as attractor-convergence (the attractor can be an equilibrium point, limit cycle or strange attractor) or oscillatory.[16]
Contemporary
Robin Holliday defined in 1990 epigenetics as "the study of the mechanisms of temporal and spatial control of gene activity during the development of complex organisms."[17] Thus, in its broadest sense, epigenetic can be used to describe anything other than DNA sequence that influences the development of an organism.
More recent usage of the word in biology follows stricter definitions. As defined by Arthur Riggs and colleagues, it is "the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence."[18]
The term has also been used, however, to describe processes which have not been demonstrated to be heritable, such as some forms of histone modification. Consequently, there are attempts to redefine "epigenetics" in broader terms that would avoid the constraints of requiring heritability. For example, Adrian Bird defined epigenetics as "the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states."[3] This definition would be inclusive of transient modifications associated with DNA repair or cell-cycle phases as well as stable changes maintained across multiple cell generations, but exclude others such as templating of membrane architecture and prions unless they impinge on chromosome function. Such redefinitions however are not universally accepted and are still subject to debate.[19] The NIH "Roadmap Epigenomics Project", ongoing as of 2016, uses the following definition: "For purposes of this program, epigenetics refers to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable."[8] In 2008, a consensus definition of the epigenetic trait, a "stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence," was made at a Cold Spring Harbor meeting.[7]
The similarity of the word to "genetics" has generated many parallel usages. The "epigenome" is a parallel to the word "genome," referring to the overall epigenetic state of a cell, and epigenomics refers to global analyses of epigenetic changes across the entire genome.[8] The phrase "genetic code" has also been adapted – the "epigenetic code" has been used to describe the set of epigenetic features that create different phenotypes in different cells from the same underlying DNA sequence. Taken to its extreme, the "epigenetic code" could represent the total state of the cell, with the position of each molecule accounted for in an epigenomic map, a diagrammatic representation of the gene expression, DNA methylation and histone modification status of a particular genomic region. More typically, the term is used in reference to systematic efforts to measure specific, relevant forms of epigenetic information such as the histone code or DNA methylation patterns.
Developmental psychology
In a sense somewhat unrelated to its use in any biological disciplines, the term "epigenetic" has also been used in developmental psychology to describe psychological development as the result of an ongoing, bi-directional interchange between heredity and the environment.[20] Interactive ideas of development have been discussed in various forms and under various names throughout the 19th and 20th centuries. An early version was proposed, among the founding statements in embryology, by Karl Ernst von Baer and popularized by Ernst Haeckel. A radical epigenetic view, known as physiological epigenesis, was developed by Paul Wintrebert. Another variation, probabilistic epigenesis, was presented by Gilbert Gottlieb in 2003.[21] This view encompasses all of the possible developing factors on an organism and how they not only influence the organism and each other but how the organism also influences its own development. Gottlieb gave an example of Rhesus monkeys where infants that did not receive typical maternal care lacked serotonin, which in turn made them more aggressive as they got older.[22] On another note, the long-standing notion "cells that fire together, wire together" derives from Hebbian theory which asserts that synaptogenesis, a developmental process with great epigenetic precedence, depends on the activity of the respective synapses within a neural network. Where experience alters the excitability of neurons, increased neural activity has been linked to increased demethylation .[23]
The developmental psychologist Erik Erikson wrote of an epigenetic principle in his 1968 book Identity: Youth and Crisis, encompassing the notion that we develop through an unfolding of our personality in predetermined stages, and that our environment and surrounding culture influence how we progress through these stages. This biological unfolding in relation to our socio-cultural settings is done in stages of psychosocial development, where "progress through each stage is in part determined by our success, or lack of success, in all the previous stages."[24][25][26]
Although empirical studies have yielded discrepant results, epigenetic modifications are thought to be a biological mechanism for transgenerational trauma.[27][28][29][30][31][32]
Molecular basis
Epigenetic changes modify the activation of certain genes, but not the genetic code sequence of DNA. The microstructure (not code) of DNA itself or the associated chromatin proteins may be modified, causing activation or silencing. This mechanism enables differentiated cells in a multicellular organism to express only the genes that are necessary for their own activity. Epigenetic changes are preserved when cells divide. Most epigenetic changes only occur within the course of one individual organism's lifetime; however, these epigenetic changes can be transmitted to the organism's offspring through a process called transgenerational epigenetic inheritance. Moreover, if gene inactivation occurs in a sperm or egg cell that results in fertilization, this epigenetic modification may also be transferred to the next generation.[33]
Specific epigenetic processes include paramutation, bookmarking, imprinting, gene silencing, X chromosome inactivation, position effect, DNA methylation reprogramming, transvection, maternal effects, the progress of carcinogenesis, many effects of teratogens, regulation of histone modifications and heterochromatin, and technical limitations affecting parthenogenesis and cloning.[34][35][36]
DNA damage
DNA damage can also cause epigenetic changes.[37][38][39] DNA damage is very frequent, occurring on average about 60,000 times a day per cell of the human body (see DNA damage (naturally occurring)). These damages are largely repaired, however, epigenetic changes can still remain at the site of DNA repair.[40] In particular, a double strand break in DNA can initiate unprogrammed epigenetic gene silencing both by causing DNA methylation as well as by promoting silencing types of histone modifications (chromatin remodeling - see next section).[41] In addition, the enzyme Parp1 (poly(ADP)-ribose polymerase) and its product poly(ADP)-ribose (PAR) accumulate at sites of DNA damage as part of the repair process.[42] This accumulation, in turn, directs recruitment and activation of the chromatin remodeling protein, ALC1, that can cause nucleosome remodeling.[43] Nucleosome remodeling has been found to cause, for instance, epigenetic silencing of DNA repair gene MLH1.[18][44] DNA damaging chemicals, such as benzene, hydroquinone, styrene, carbon tetrachloride and trichloroethylene, cause considerable hypomethylation of DNA, some through the activation of oxidative stress pathways.[45]
Foods are known to alter the epigenetics of rats on different diets.[46] Some food components epigenetically increase the levels of DNA repair enzymes such as MGMT and MLH1[47] and p53.[48][49] Other food components can reduce DNA damage, such as soy isoflavones. In one study, markers for oxidative stress, such as modified nucleotides that can result from DNA damage, were decreased by a 3-week diet supplemented with soy.[50] A decrease in oxidative DNA damage was also observed 2 h after consumption of anthocyanin-rich bilberry (Vaccinium myrtillius L.) pomace extract.[51]
Techniques used to study epigenetics
Epigenetic research uses a wide range of molecular biological techniques to further understanding of epigenetic phenomena. These techniques include chromatin immunoprecipitation (together with its large-scale variants ChIP-on-chip and ChIP-Seq), fluorescent in situ hybridization, methylation-sensitive restriction enzymes, DNA adenine methyltransferase identification (DamID) and bisulfite sequencing.[52] Furthermore, the use of bioinformatics methods has a role in computational epigenetics.[52]
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) has helped bridge the gap between DNA and epigenetic interactions.[53] With the use of ChIP, researchers are able to make findings in regards to gene regulation, transcription mechanisms, and chromatin structure.[53]
Fluorescent in situ hybridization
Fluorescent in situ hybridization (FISH) is very important to understand epigenetic mechanisms.[54] FISH can be used to find the location of genes on chromosomes, as well as finding noncoding RNAs.[54][55] FISH is predominantly used for detecting chromosomal abnormalities in humans.[55]
Methylation-sensitive restriction enzymes
Methylation sensitive restriction enzymes paired with PCR is a way to evaluate methylation in DNA - specifically the CpG sites.[56] If DNA is methylated, the restriction enzymes will not cleave the strand.[56] Contrarily, if the DNA is not methylated, the enzymes will cleave the strand and it will be amplified by PCR.[56]
Bisulfite sequencing
Bisulfite sequencing is another way to evaluate DNA methylation. Cytosine will be changed to uracil from being treated with sodium bisulfite, whereas methylated cytosines will not be affected.[56]
Mechanisms
Epigenetic mechanisms are sensitive to environmental effects, and key participants in shaping an adult phenotype.[57] Several types of epigenetic inheritance systems may play a role in what has become known as cell memory,[58] note however that not all of these are universally accepted to be examples of epigenetics.
Covalent modifications
Covalent modification of either DNA (e.g. cytosine methylation and hydroxymethylation) or of histone proteins (e.g. lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, and lysine ubiquitination and sumoylation) play central roles in many types of epigenetic inheritance. Therefore, the word "epigenetics" is sometimes used as a synonym for these processes. However, this can be misleading. Chromatin remodeling is not always inherited, and not all epigenetic inheritance involves chromatin remodeling.[59] In 2019, a further lysine modification appeared in the scientific literature linking epigenetics modification to cell metabolism, i.e. Lactylation[60]
Because the phenotype of a cell or individual is affected by which of its genes are transcribed, heritable transcription states can give rise to epigenetic effects. There are several layers of regulation of gene expression. One way that genes are regulated is through the remodeling of chromatin. Chromatin is the complex of DNA and the histone proteins with which it associates. If the way that DNA is wrapped around the histones changes, gene expression can change as well. Chromatin remodeling is accomplished through two main mechanisms:
- The first way is post translational modification of the amino acids that make up histone proteins. Histone proteins are made up of long chains of amino acids. If the amino acids that are in the chain are changed, the shape of the histone might be modified. DNA is not completely unwound during replication. It is possible, then, that the modified histones may be carried into each new copy of the DNA. Once there, these histones may act as templates, initiating the surrounding new histones to be shaped in the new manner. By altering the shape of the histones around them, these modified histones would ensure that a lineage-specific transcription program is maintained after cell division.
- The second way is the addition of methyl groups to the DNA, mostly at CpG sites, to convert cytosine to 5-methylcytosine. 5-Methylcytosine performs much like a regular cytosine, pairing with a guanine in double-stranded DNA. However, when methylated cytosines are present in CpG sites in the promoter and enhancer regions of genes, the genes are often repressed.[61][62] When methylated cytosines are present in CpG sites in the gene body (in the coding region excluding the transcription start site) expression of the gene is often enhanced. Transcription of a gene usually depends on a transcription factor binding to a (10 base or less) recognition sequence at the enhancer that interacts with the promoter region of that gene (Gene expression # Enhancers, transcription factors, mediator complex and DNA loops in mammalian transcription).[63] About 22% of transcription factors are inhibited from binding when the recognition sequence has a methylated cytosine. In addition, presence of methylated cytosines at a promoter region can attract methyl-CpG-binding domain (MBD) proteins. All MBDs interact with nucleosome remodeling and histone deacetylase complexes, which leads to gene silencing. In addition, another covalent modification involving methylated cytosine is its demethylation by TET enzymes. Hundreds of such demethylations occur, for instance, during learning and memory forming events in neurons.[64][65]
There is frequently a reciprocal relationship between DNA methylation and histone lysine methylation.[66] For instance, the methyl binding domain protein MBD1, attracted to and associating with methylated cytosine in a DNA CpG site, can also associate with H3K9 methyltransferase activity to methylate histone 3 at lysine 9. On the other hand, DNA maintenance methylation by DNMT1 appears to partly rely on recognition of histone methylation on the nucleosome present at the DNA site to carry out cytosine methylation on newly synthesized DNA.[66] There is further crosstalk between DNA methylation carried out by DNMT3A and DNMT3B and histone methylation so that there is a correlation between the genome-wide distribution of DNA methylation and histone methylation.[67]
Mechanisms of heritability of histone state are not well understood; however, much is known about the mechanism of heritability of DNA methylation state during cell division and differentiation. Heritability of methylation state depends on certain enzymes (such as DNMT1) that have a higher affinity for 5-methylcytosine than for cytosine. If this enzyme reaches a "hemimethylated" portion of DNA (where 5-methylcytosine is in only one of the two DNA strands) the enzyme will methylate the other half. However, it is now known that DNMT1 physically interacts with the protein UHRF1. UHRF1 has been recently recognized as essential for DNMT1-mediated maintenance of DNA methylation. UHRF1 is the protein that specifically recognizes hemi-methylated DNA, therefore bringing DNMT1 to its substrate to maintain DNA methylation.[67]
Although histone modifications occur throughout the entire sequence, the unstructured N-termini of histones (called histone tails) are particularly highly modified. These modifications include acetylation, methylation, ubiquitylation, phosphorylation, sumoylation, ribosylation and citrullination. Acetylation is the most highly studied of these modifications. For example, acetylation of the K14 and K9 lysines of the tail of histone H3 by histone acetyltransferase enzymes (HATs) is generally related to transcriptional competence[69] (see Figure).
One mode of thinking is that this tendency of acetylation to be associated with "active" transcription is biophysical in nature. Because it normally has a positively charged nitrogen at its end, lysine can bind the negatively charged phosphates of the DNA backbone. The acetylation event converts the positively charged amine group on the side chain into a neutral amide linkage. This removes the positive charge, thus loosening the DNA from the histone. When this occurs, complexes like SWI/SNF and other transcriptional factors can bind to the DNA and allow transcription to occur. This is the "cis" model of the epigenetic function. In other words, changes to the histone tails have a direct effect on the DNA itself. [70]
Another model of epigenetic function is the "trans" model. In this model, changes to the histone tails act indirectly on the DNA. For example, lysine acetylation may create a binding site for chromatin-modifying enzymes (or transcription machinery as well). This chromatin remodeler can then cause changes to the state of the chromatin. Indeed, a bromodomain – a protein domain that specifically binds acetyl-lysine – is found in many enzymes that help activate transcription, including the SWI/SNF complex. It may be that acetylation acts in this and the previous way to aid in transcriptional activation.
The idea that modifications act as docking modules for related factors is borne out by histone methylation as well. Methylation of lysine 9 of histone H3 has long been associated with constitutively transcriptionally silent chromatin (constitutive heterochromatin) (see bottom Figure). It has been determined that a chromodomain (a domain that specifically binds methyl-lysine) in the transcriptionally repressive protein HP1 recruits HP1 to K9 methylated regions. One example that seems to refute this biophysical model for methylation is that tri-methylation of histone H3 at lysine 4 is strongly associated with (and required for full) transcriptional activation (see top Figure). Tri-methylation, in this case, would introduce a fixed positive charge on the tail.
It has been shown that the histone lysine methyltransferase (KMT) is responsible for this methylation activity in the pattern of histones H3 & H4. This enzyme utilizes a catalytically active site called the SET domain (Suppressor of variegation, Enhancer of Zeste, Trithorax). The SET domain is a 130-amino acid sequence involved in modulating gene activities. This domain has been demonstrated to bind to the histone tail and causes the methylation of the histone.[71]
Differing histone modifications are likely to function in differing ways; acetylation at one position is likely to function differently from acetylation at another position. Also, multiple modifications may occur at the same time, and these modifications may work together to change the behavior of the nucleosome. The idea that multiple dynamic modifications regulate gene transcription in a systematic and reproducible way is called the histone code, although the idea that histone state can be read linearly as a digital information carrier has been largely debunked. One of the best-understood systems that orchestrate chromatin-based silencing is the SIR protein based silencing of the yeast hidden mating-type loci HML and HMR.
DNA methylation frequently occurs in repeated sequences, and helps to suppress the expression and mobility of 'transposable elements':[72] Because 5-methylcytosine can be spontaneously deaminated (replacing nitrogen by oxygen) to thymidine, CpG sites are frequently mutated and become rare in the genome, except at CpG islands where they remain unmethylated. Epigenetic changes of this type thus have the potential to direct increased frequencies of permanent genetic mutation. DNA methylation patterns are known to be established and modified in response to environmental factors by a complex interplay of at least three independent DNA methyltransferases, DNMT1, DNMT3A, and DNMT3B, the loss of any of which is lethal in mice.[73] DNMT1 is the most abundant methyltransferase in somatic cells,[74] localizes to replication foci,[75] has a 10–40-fold preference for hemimethylated DNA and interacts with the proliferating cell nuclear antigen (PCNA).[76]
By preferentially modifying hemimethylated DNA, DNMT1 transfers patterns of methylation to a newly synthesized strand after DNA replication, and therefore is often referred to as the ‘maintenance' methyltransferase.[77] DNMT1 is essential for proper embryonic development, imprinting and X-inactivation.[73][78] To emphasize the difference of this molecular mechanism of inheritance from the canonical Watson-Crick base-pairing mechanism of transmission of genetic information, the term 'Epigenetic templating' was introduced.[79] Furthermore, in addition to the maintenance and transmission of methylated DNA states, the same principle could work in the maintenance and transmission of histone modifications and even cytoplasmic (structural) heritable states.[80]
Histones H3 and H4 can also be manipulated through demethylation using histone lysine demethylase (KDM). This recently identified enzyme has a catalytically active site called the Jumonji domain (JmjC). The demethylation occurs when JmjC utilizes multiple cofactors to hydroxylate the methyl group, thereby removing it. JmjC is capable of demethylating mono-, di-, and tri-methylated substrates.[81]
Chromosomal regions can adopt stable and heritable alternative states resulting in bistable gene expression without changes to the DNA sequence. Epigenetic control is often associated with alternative covalent modifications of histones.[82] The stability and heritability of states of larger chromosomal regions are suggested to involve positive feedback where modified nucleosomes recruit enzymes that similarly modify nearby nucleosomes.[83] A simplified stochastic model for this type of epigenetics is found here.[84][85]
It has been suggested that chromatin-based transcriptional regulation could be mediated by the effect of small RNAs. Small interfering RNAs can modulate transcriptional gene expression via epigenetic modulation of targeted promoters.[86]
RNA transcripts
Sometimes a gene, after being turned on, transcribes a product that (directly or indirectly) maintains the activity of that gene. For example, Hnf4 and MyoD enhance the transcription of many liver-specific and muscle-specific genes, respectively, including their own, through the transcription factor activity of the proteins they encode. RNA signalling includes differential recruitment of a hierarchy of generic chromatin modifying complexes and DNA methyltransferases to specific loci by RNAs during differentiation and development.[87] Other epigenetic changes are mediated by the production of different splice forms of RNA, or by formation of double-stranded RNA (RNAi). Descendants of the cell in which the gene was turned on will inherit this activity, even if the original stimulus for gene-activation is no longer present. These genes are often turned on or off by signal transduction, although in some systems where syncytia or gap junctions are important, RNA may spread directly to other cells or nuclei by diffusion. A large amount of RNA and protein is contributed to the zygote by the mother during oogenesis or via nurse cells, resulting in maternal effect phenotypes. A smaller quantity of sperm RNA is transmitted from the father, but there is recent evidence that this epigenetic information can lead to visible changes in several generations of offspring.[88]
MicroRNAs
MicroRNAs (miRNAs) are members of non-coding RNAs that range in size from 17 to 25 nucleotides. miRNAs regulate a large variety of biological functions in plants and animals.[89] So far, in 2013, about 2000 miRNAs have been discovered in humans and these can be found online in a miRNA database.[90] Each miRNA expressed in a cell may target about 100 to 200 messenger RNAs(mRNAs) that it downregulates.[91] Most of the downregulation of mRNAs occurs by causing the decay of the targeted mRNA, while some downregulation occurs at the level of translation into protein.[92]
It appears that about 60% of human protein coding genes are regulated by miRNAs.[93] Many miRNAs are epigenetically regulated. About 50% of miRNA genes are associated with CpG islands,[89] that may be repressed by epigenetic methylation. Transcription from methylated CpG islands is strongly and heritably repressed.[94] Other miRNAs are epigenetically regulated by either histone modifications or by combined DNA methylation and histone modification.[89]
mRNA
In 2011, it was demonstrated that the methylation of mRNA plays a critical role in human energy homeostasis. The obesity-associated FTO gene is shown to be able to demethylate N6-methyladenosine in RNA.[95][96]
sRNAs
sRNAs are small (50–250 nucleotides), highly structured, non-coding RNA fragments found in bacteria. They control gene expression including virulence genes in pathogens and are viewed as new targets in the fight against drug-resistant bacteria.[97] They play an important role in many biological processes, binding to mRNA and protein targets in prokaryotes. Their phylogenetic analyses, for example through sRNA–mRNA target interactions or protein binding properties, are used to build comprehensive databases.[98] sRNA-gene maps based on their targets in microbial genomes are also constructed.[99]
Prions
Prions are infectious forms of proteins. In general, proteins fold into discrete units that perform distinct cellular functions, but some proteins are also capable of forming an infectious conformational state known as a prion. Although often viewed in the context of infectious disease, prions are more loosely defined by their ability to catalytically convert other native state versions of the same protein to an infectious conformational state. It is in this latter sense that they can be viewed as epigenetic agents capable of inducing a phenotypic change without a modification of the genome.[100]
Fungal prions are considered by some to be epigenetic because the infectious phenotype caused by the prion can be inherited without modification of the genome. PSI+ and URE3, discovered in yeast in 1965 and 1971, are the two best studied of this type of prion.[101][102] Prions can have a phenotypic effect through the sequestration of protein in aggregates, thereby reducing that protein's activity. In PSI+ cells, the loss of the Sup35 protein (which is involved in termination of translation) causes ribosomes to have a higher rate of read-through of stop codons, an effect that results in suppression of nonsense mutations in other genes.[103] The ability of Sup35 to form prions may be a conserved trait. It could confer an adaptive advantage by giving cells the ability to switch into a PSI+ state and express dormant genetic features normally terminated by stop codon mutations.[104][105][106][107]
Prion-based epigenetics has also been observed in Saccharomyces cerevisiae.[108]
Structural inheritance
In ciliates such as Tetrahymena and Paramecium, genetically identical cells show heritable differences in the patterns of ciliary rows on their cell surface. Experimentally altered patterns can be transmitted to daughter cells. It seems existing structures act as templates for new structures. The mechanisms of such inheritance are unclear, but reasons exist to assume that multicellular organisms also use existing cell structures to assemble new ones.[109][110][111]
Nucleosome positioning
Eukaryotic genomes have numerous nucleosomes. Nucleosome position is not random, and determine the accessibility of DNA to regulatory proteins. Promoters active in different tissues have been shown to have different nucleosome positioning features.[112] This determines differences in gene expression and cell differentiation. It has been shown that at least some nucleosomes are retained in sperm cells (where most but not all histones are replaced by protamines). Thus nucleosome positioning is to some degree inheritable. Recent studies have uncovered connections between nucleosome positioning and other epigenetic factors, such as DNA methylation and hydroxymethylation.[113]
Histone variants
Different histone variants are incorporated into specific regions of the genome non-randomly. Their differential biochemical characteristics can affect genome functions via their roles in gene regulation,[114] and maintenance of chromosome structures.[115]
Genomic architecture
The three-dimensional configuration of the genome (the 3D genome) is complex, dynamic and crucial for regulating genomic function and nuclear processes such as DNA replication, transcription and DNA-damage repair.[116]
Functions and consequences
Memory
Memory formation and maintenance are due to epigenetic alterations that cause the required dynamic changes in gene transcription that create and renew memory in neurons.[117]
An event can set off a chain of reactions that result in altered methylations of a large set of genes in neurons, which give a representation of the event, a memory.[117]
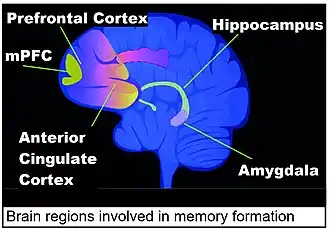
Areas of the brain important in the formation of memories include the hippocampus, medial prefrontal cortex (mPFC), anterior cingulate cortex and amygdala, as shown in the diagram of the human brain in this section.[118]
When a strong memory is created, as in a rat subjected to contextual fear conditioning (CFC), one of the earliest events to occur is that more than 100 DNA double-strand breaks are formed by topoisomerase IIB in neurons of the hippocampus and the medial prefrontal cortex (mPFC).[119] These double-strand breaks are at specific locations that allow activation of transcription of immediate early genes (IEGs) that are important in memory formation, allowing their expression in mRNA, with peak mRNA transcription at seven to ten minutes after CFC.[119][120]
Two important IEGs in memory formation are EGR1[121] and the alternative promoter variant of DNMT3A, DNMT3A2.[122] EGR1 protein binds to DNA at its binding motifs, 5′-GCGTGGGCG-3′ or 5′-GCGGGGGCGG-3', and there are about 12,000 genome locations at which EGR1 protein can bind.[123] EGR1 protein binds to DNA in gene promoter and enhancer regions. EGR1 recruits the demethylating enzyme TET1 to an association, and brings TET1 to about 600 locations on the genome where TET1 can then demethylate and activate the associated genes.[123]
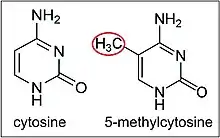
The DNA methyltransferases DNMT3A1, DNMT3A2 and DNMT3B can all methylate cytosines (see image this section) at CpG sites in or near the promoters of genes. As shown by Manzo et al., [124] these three DNA methyltransferases differ in their genomic binding locations and DNA methylation activity at different regulatory sites. Manzo et al. located 3,970 genome regions exclusively enriched for DNMT3A1, 3,838 regions for DNMT3A2 and 3,432 regions for DNMT3B. When DNMT3A2 is newly induced as an IEG (when neurons are activated), many new cytosine methylations occur, presumably in the target regions of DNMT3A2. Oliviera et al.[122] found that the neuronal activity-inducible IEG levels of Dnmt3a2 in the hippocampus determined the ability to form long-term memories.
Rats form long-term associative memories after contextual fear conditioning (CFC).[125] Duke et al.[64] found that 24 hours after CFC in rats, in hippocampus neurons, 2,097 genes (9.17% of the genes in the rat genome) had altered methylation. When newly methylated cytosines are present in CpG sites in the promoter regions of genes, the genes are often repressed, and when newly demethylated cytosines are present the genes may be activated.[126] After CFC, there were 1,048 genes with reduced mRNA expression and 564 genes with upregulated mRNA expression. Similarly, when mice undergo CFC, one hour later in the hippocampus region of the mouse brain there are 675 demethylated genes and 613 hypermethylated genes.[127] However, memories do not remain in the hippocampus, but after four or five weeks the memories are stored in the anterior cingulate cortex.[128] In the studies on mice after CFC, Halder et al.[127] showed that four weeks after CFC there were at least 1,000 differentially methylated genes and more than 1,000 differentially expressed genes in the anterior cingulate cortex, while at the same time the altered methylations in the hippocampus were reversed.
The epigenetic alteration of methylation after a new memory is established creates a different pool of nuclear mRNAs. As reviewed by Bernstein,[117] the epigenetically determined new mix of nuclear mRNAs are often packaged into neuronal granules, or messenger RNP, consisting of mRNA, small and large ribosomal subunits, translation initiation factors and RNA-binding proteins that regulate mRNA function. These neuronal granules are transported from the neuron nucleus and are directed, according to 3′ untranslated regions of the mRNA in the granules (their "zip codes"), to neuronal dendrites. Roughly 2,500 mRNAs may be localized to the dendrites of hippocampal pyramidal neurons and perhaps 450 transcripts are in excitatory presynaptic nerve terminals (dendritic spines). The altered assortments of transcripts (dependent on epigenetic alterations in the neuron nucleus) have different sensitivities in response to signals, which is the basis of altered synaptic plasticity. Altered synaptic plasticity is often considered the neurochemical foundation of learning and memory.
Development
Developmental epigenetics can be divided into predetermined and probabilistic epigenesis. Predetermined epigenesis is a unidirectional movement from structural development in DNA to the functional maturation of the protein. "Predetermined" here means that development is scripted and predictable. Probabilistic epigenesis on the other hand is a bidirectional structure-function development with experiences and external molding development.[129]
Somatic epigenetic inheritance, particularly through DNA and histone covalent modifications and nucleosome repositioning, is very important in the development of multicellular eukaryotic organisms.[113] The genome sequence is static (with some notable exceptions), but cells differentiate into many different types, which perform different functions, and respond differently to the environment and intercellular signaling. Thus, as individuals develop, morphogens activate or silence genes in an epigenetically heritable fashion, giving cells a memory. In mammals, most cells terminally differentiate, with only stem cells retaining the ability to differentiate into several cell types ("totipotency" and "multipotency"). In mammals, some stem cells continue producing newly differentiated cells throughout life, such as in neurogenesis, but mammals are not able to respond to loss of some tissues, for example, the inability to regenerate limbs, which some other animals are capable of. Epigenetic modifications regulate the transition from neural stem cells to glial progenitor cells (for example, differentiation into oligodendrocytes is regulated by the deacetylation and methylation of histones.[130] Unlike animals, plant cells do not terminally differentiate, remaining totipotent with the ability to give rise to a new individual plant. While plants do utilize many of the same epigenetic mechanisms as animals, such as chromatin remodeling, it has been hypothesized that some kinds of plant cells do not use or require "cellular memories", resetting their gene expression patterns using positional information from the environment and surrounding cells to determine their fate.[131]
Epigenetic changes can occur in response to environmental exposure – for example, maternal dietary supplementation with genistein (250 mg/kg) have epigenetic changes affecting expression of the agouti gene, which affects their fur color, weight, and propensity to develop cancer.[132][133][134] Ongoing research is focused on exploring the impact of other known teratogens, such as diabetic embryopathy, on methylation signatures.[135]
Controversial results from one study suggested that traumatic experiences might produce an epigenetic signal that is capable of being passed to future generations. Mice were trained, using foot shocks, to fear a cherry blossom odor. The investigators reported that the mouse offspring had an increased aversion to this specific odor.[136][137] They suggested epigenetic changes that increase gene expression, rather than in DNA itself, in a gene, M71, that governs the functioning of an odor receptor in the nose that responds specifically to this cherry blossom smell. There were physical changes that correlated with olfactory (smell) function in the brains of the trained mice and their descendants. Several criticisms were reported, including the study's low statistical power as evidence of some irregularity such as bias in reporting results.[138] Due to limits of sample size, there is a probability that an effect will not be demonstrated to within statistical significance even if it exists. The criticism suggested that the probability that all the experiments reported would show positive results if an identical protocol was followed, assuming the claimed effects exist, is merely 0.4%. The authors also did not indicate which mice were siblings, and treated all of the mice as statistically independent.[139] The original researchers pointed out negative results in the paper's appendix that the criticism omitted in its calculations, and undertook to track which mice were siblings in the future.[140]
Transgenerational
Epigenetic mechanisms were a necessary part of the evolutionary origin of cell differentiation.[141] Although epigenetics in multicellular organisms is generally thought to be a mechanism involved in differentiation, with epigenetic patterns "reset" when organisms reproduce, there have been some observations of transgenerational epigenetic inheritance (e.g., the phenomenon of paramutation observed in maize). Although most of these multigenerational epigenetic traits are gradually lost over several generations, the possibility remains that multigenerational epigenetics could be another aspect to evolution and adaptation. As mentioned above, some define epigenetics as heritable.
A sequestered germ line or Weismann barrier is specific to animals, and epigenetic inheritance is more common in plants and microbes. Eva Jablonka, Marion J. Lamb and Étienne Danchin have argued that these effects may require enhancements to the standard conceptual framework of the modern synthesis and have called for an extended evolutionary synthesis.[142][143][144] Other evolutionary biologists, such as John Maynard Smith, have incorporated epigenetic inheritance into population-genetics models[145] or are openly skeptical of the extended evolutionary synthesis (Michael Lynch).[146] Thomas Dickins and Qazi Rahman state that epigenetic mechanisms such as DNA methylation and histone modification are genetically inherited under the control of natural selection and therefore fit under the earlier "modern synthesis".[147]
Two important ways in which epigenetic inheritance can differ from traditional genetic inheritance, with important consequences for evolution, are:
- rates of epimutation can be much faster than rates of mutation[148]
- the epimutations are more easily reversible[149]
In plants, heritable DNA methylation mutations are 100,000 times more likely to occur compared to DNA mutations.[150] An epigenetically inherited element such as the PSI+ system can act as a "stop-gap", good enough for short-term adaptation that allows the lineage to survive for long enough for mutation and/or recombination to genetically assimilate the adaptive phenotypic change.[151] The existence of this possibility increases the evolvability of a species.
More than 100 cases of transgenerational epigenetic inheritance phenomena have been reported in a wide range of organisms, including prokaryotes, plants, and animals.[152] For instance, mourning-cloak butterflies will change color through hormone changes in response to experimentation of varying temperatures.[153]
The filamentous fungus Neurospora crassa is a prominent model system for understanding the control and function of cytosine methylation. In this organism, DNA methylation is associated with relics of a genome-defense system called RIP (repeat-induced point mutation) and silences gene expression by inhibiting transcription elongation.[154]
The yeast prion PSI is generated by a conformational change of a translation termination factor, which is then inherited by daughter cells. This can provide a survival advantage under adverse conditions, exemplifying epigenetic regulation which enables unicellular organisms to respond rapidly to environmental stress. Prions can be viewed as epigenetic agents capable of inducing a phenotypic change without modification of the genome.[155]
Direct detection of epigenetic marks in microorganisms is possible with single molecule real time sequencing, in which polymerase sensitivity allows for measuring methylation and other modifications as a DNA molecule is being sequenced.[156] Several projects have demonstrated the ability to collect genome-wide epigenetic data in bacteria.[157][158][159][160]
Epigenetics in bacteria
While epigenetics is of fundamental importance in eukaryotes, especially metazoans, it plays a different role in bacteria.[161] Most importantly, eukaryotes use epigenetic mechanisms primarily to regulate gene expression which bacteria rarely do. However, bacteria make widespread use of postreplicative DNA methylation for the epigenetic control of DNA-protein interactions. Bacteria also use DNA adenine methylation (rather than DNA cytosine methylation) as an epigenetic signal. DNA adenine methylation is important in bacteria virulence in organisms such as Escherichia coli, Salmonella, Vibrio, Yersinia, Haemophilus, and Brucella. In Alphaproteobacteria, methylation of adenine regulates the cell cycle and couples gene transcription to DNA replication. In Gammaproteobacteria, adenine methylation provides signals for DNA replication, chromosome segregation, mismatch repair, packaging of bacteriophage, transposase activity and regulation of gene expression.[155][162] There exists a genetic switch controlling Streptococcus pneumoniae (the pneumococcus) that allows the bacterium to randomly change its characteristics into six alternative states that could pave the way to improved vaccines. Each form is randomly generated by a phase variable methylation system. The ability of the pneumococcus to cause deadly infections is different in each of these six states. Similar systems exist in other bacterial genera.[163] In Bacillota such as Clostridioides difficile, adenine methylation regulates sporulation, biofilm formation and host-adaptation.[164]
Medicine
Epigenetics has many and varied potential medical applications.[165] In 2008, the National Institutes of Health announced that $190 million had been earmarked for epigenetics research over the next five years. In announcing the funding, government officials noted that epigenetics has the potential to explain mechanisms of aging, human development, and the origins of cancer, heart disease, mental illness, as well as several other conditions. Some investigators, like Randy Jirtle, Ph.D., of Duke University Medical Center, think epigenetics may ultimately turn out to have a greater role in disease than genetics.[166]
Twins
Direct comparisons of identical twins constitute an optimal model for interrogating environmental epigenetics. In the case of humans with different environmental exposures, monozygotic (identical) twins were epigenetically indistinguishable during their early years, while older twins had remarkable differences in the overall content and genomic distribution of 5-methylcytosine DNA and histone acetylation.[6] The twin pairs who had spent less of their lifetime together and/or had greater differences in their medical histories were those who showed the largest differences in their levels of 5-methylcytosine DNA and acetylation of histones H3 and H4.[167]
Dizygotic (fraternal) and monozygotic (identical) twins show evidence of epigenetic influence in humans.[167][168][169] DNA sequence differences that would be abundant in a singleton-based study do not interfere with the analysis. Environmental differences can produce long-term epigenetic effects, and different developmental monozygotic twin subtypes may be different with respect to their susceptibility to be discordant from an epigenetic point of view.[170]
A high-throughput study, which denotes technology that looks at extensive genetic markers, focused on epigenetic differences between monozygotic twins to compare global and locus-specific changes in DNA methylation and histone modifications in a sample of 40 monozygotic twin pairs.[167] In this case, only healthy twin pairs were studied, but a wide range of ages was represented, between 3 and 74 years. One of the major conclusions from this study was that there is an age-dependent accumulation of epigenetic differences between the two siblings of twin pairs. This accumulation suggests the existence of epigenetic "drift". Epigenetic drift is the term given to epigenetic modifications as they occur as a direct function with age. While age is a known risk factor for many diseases, age-related methylation has been found to occur differentially at specific sites along the genome. Over time, this can result in measurable differences between biological and chronological age. Epigenetic changes have been found to be reflective of lifestyle and may act as functional biomarkers of disease before clinical threshold is reached.[171]
A more recent study, where 114 monozygotic twins and 80 dizygotic twins were analyzed for the DNA methylation status of around 6000 unique genomic regions, concluded that epigenetic similarity at the time of blastocyst splitting may also contribute to phenotypic similarities in monozygotic co-twins. This supports the notion that microenvironment at early stages of embryonic development can be quite important for the establishment of epigenetic marks.[168] Congenital genetic disease is well understood and it is clear that epigenetics can play a role, for example, in the case of Angelman syndrome and Prader-Willi syndrome. These are normal genetic diseases caused by gene deletions or inactivation of the genes but are unusually common because individuals are essentially hemizygous because of genomic imprinting, and therefore a single gene knock out is sufficient to cause the disease, where most cases would require both copies to be knocked out.[172]
Genomic imprinting
Some human disorders are associated with genomic imprinting, a phenomenon in mammals where the father and mother contribute different epigenetic patterns for specific genomic loci in their germ cells.[173] The best-known case of imprinting in human disorders is that of Angelman syndrome and Prader-Willi syndrome – both can be produced by the same genetic mutation, chromosome 15q partial deletion, and the particular syndrome that will develop depends on whether the mutation is inherited from the child's mother or from their father.[174] This is due to the presence of genomic imprinting in the region. Beckwith-Wiedemann syndrome is also associated with genomic imprinting, often caused by abnormalities in maternal genomic imprinting of a region on chromosome 11.
Methyl CpG-binding protein 2 (MeCP2) is a transcriptional regulator that must be phosphorylated before releasing from the BDNF promoter, allowing transcription. Rett syndrome is underlain by mutations in the MeCP2 gene despite no large-scale changes in expression of MeCP2 being found in microarray analyses. BDNF is downregulated in the MECP2 mutant resulting in Rett syndrome, as well as the increase of early neural senescence and accumulation of damaged DNA.[175]
In the Överkalix study, paternal (but not maternal) grandsons[176] of Swedish men who were exposed during preadolescence to famine in the 19th century were less likely to die of cardiovascular disease. If food was plentiful, then diabetes mortality in the grandchildren increased, suggesting that this was a transgenerational epigenetic inheritance.[177] The opposite effect was observed for females – the paternal (but not maternal) granddaughters of women who experienced famine while in the womb (and therefore while their eggs were being formed) lived shorter lives on average.[178]
Diabetic wound healing
Epigenetic modifications have given insight into the understanding of the pathophysiology of different disease conditions. Though, they are strongly associated with cancer, their role in other pathological conditions are of equal importance. It appears that the hyperglycaemic environment could imprint such changes at the genomic level, that macrophages are primed towards a pro-inflammatory state and could fail to exhibit any phenotypic alteration towards the pro-healing type. This phenomenon of altered Macrophage Polarization is mostly associated with all the diabetic complications in a clinical set-up. As of 2018, several reports reveal the relevance of different epigenetic modifications with respect to diabetic complications. Sooner or later, with the advancements in biomedical tools, the detection of such biomarkers as prognostic and diagnostic tools in patients could possibly emerge out as alternative approaches. It is noteworthy to mention here that the use of epigenetic modifications as therapeutic targets warrant extensive preclinical as well as clinical evaluation prior to use.[179]
Examples of drugs altering gene expression from epigenetic events
The use of beta-lactam antibiotics can alter glutamate receptor activity and the action of cyclosporine on multiple transcription factors. Additionally, lithium can impact autophagy of aberrant proteins, and opioid drugs via chronic use can increase the expression of genes associated with addictive phenotypes.[180]
In a groundbreaking 2003 report, Caspi and colleagues demonstrated that in a robust cohort of over one-thousand subjects assessed multiple times from preschool to adulthood, subjects who carried one or two copies of the short allele of the serotonin transporter promoter polymorphism exhibited higher rates of adult depression and suicidality when exposed to childhood maltreatment when compared to long allele homozygotes with equal ELS exposure.[181]
Parental nutrition, in utero exposure to stress or endocrine disrupting chemicals,[182] male-induced maternal effects such as the attraction of differential mate quality, and maternal as well as paternal age, and offspring gender could all possibly influence whether a germline epimutation is ultimately expressed in offspring and the degree to which intergenerational inheritance remains stable throughout posterity.[183] However, whether and to what extent epigenetic effects can be transmitted across generations remains unclear, particularly in humans.[184][185]
Addiction
Addiction is a disorder of the brain's reward system which arises through transcriptional and neuroepigenetic mechanisms and occurs over time from chronically high levels of exposure to an addictive stimulus (e.g., morphine, cocaine, sexual intercourse, gambling, etc.).[186][187][188][189] Transgenerational epigenetic inheritance of addictive phenotypes has been noted to occur in preclinical studies.[190][191] However, robust evidence in support of the persistence of epigenetic effects across multiple generations has yet to be established in humans; for example, an epigenetic effect of prenatal exposure to smoking that is observed in great-grandchildren who had not been exposed.[184]
Depression
Epigenetic inheritance of depression-related phenotypes has also been reported in a preclinical study.[192] Inheritance of paternal stress-induced traits across generations involved small non-coding RNA signals transmitted via the paternal germline.
Research
The two forms of heritable information, namely genetic and epigenetic, are collectively called dual inheritance. Members of the APOBEC/AID family of cytosine deaminases may concurrently influence genetic and epigenetic inheritance using similar molecular mechanisms, and may be a point of crosstalk between these conceptually compartmentalized processes.[193]
Fluoroquinolone antibiotics induce epigenetic changes in mammalian cells through iron chelation. This leads to epigenetic effects through inhibition of α-ketoglutarate-dependent dioxygenases that require iron as a co-factor.[194]
Various pharmacological agents are applied for the production of induced pluripotent stem cells (iPSC) or maintain the embryonic stem cell (ESC) phenotypic via epigenetic approach. Adult stem cells like bone marrow stem cells have also shown a potential to differentiate into cardiac competent cells when treated with G9a histone methyltransferase inhibitor BIX01294.[195][196]
Pseudoscience
As epigenetics is in the early stages of development as a science and is surrounded by sensationalism in the public media, David Gorski and geneticist Adam Rutherford have advised caution against the proliferation of false and pseudoscientific conclusions by new age authors making unfounded suggestions that a person's genes and health can be manipulated by mind control. Misuse of the scientific term by quack authors has produced misinformation among the general public.[2][197]
See also
- Baldwin effect
- Behavioral epigenetics
- Biological effects of radiation on the epigenome
- Computational epigenetics
- Contribution of epigenetic modifications to evolution
- DAnCER database (2010)
- Epigenesis (biology)
- Epigenetics in forensic science
- Epigenetic therapy
- Epigenetics of neurodegenerative diseases
- Genetics
- Lamarckism
- Nutriepigenomics
- Position-effect variegation
- Preformationism
- Somatic epitype
- Synthetic genetic array
- Transcriptional memory
- Transgenerational epigenetic inheritance
References
- Dupont C, Armant DR, Brenner CA (September 2009). "Epigenetics: definition, mechanisms and clinical perspective". Seminars in Reproductive Medicine. 27 (5): 351–7. doi:10.1055/s-0029-1237423. PMC 2791696. PMID 19711245.
In the original sense of this definition, epigenetics referred to all molecular pathways modulating the expression of a genotype into a particular phenotype. Over the following years, with the rapid growth of genetics, the meaning of the word has gradually narrowed. Epigenetics has been defined and today is generally accepted as 'the study of changes in gene function that are mitotically and/or meiotically heritable and that do not entail a change in DNA sequence.'
- Rutherford A (19 July 2015). "Beware the pseudo gene genies". The Guardian.
- Bird A (May 2007). "Perceptions of epigenetics". Nature. 447 (7143): 396–8. Bibcode:2007Natur.447..396B. doi:10.1038/nature05913. PMID 17522671. S2CID 4357965.
- Hunter P (1 May 2008). "What genes remember". Prospect Magazine. Archived from the original on 1 May 2008. Retrieved 26 July 2012.
- Reik W (May 2007). "Stability and flexibility of epigenetic gene regulation in mammalian development". Nature. 447 (7143): 425–32. Bibcode:2007Natur.447..425R. doi:10.1038/nature05918. PMID 17522676. S2CID 11794102.
- Moore DS (2015). The Developing Genome: An Introduction to Behavioral Epigenetics (1st ed.). Oxford University Press. ISBN 978-0199922345.
- Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A (April 2009). "An operational definition of epigenetics". Genes & Development. 23 (7): 781–3. doi:10.1101/gad.1787609. PMC 3959995. PMID 19339683.
- "Overview". NIH Roadmap Epigenomics Project.
- Oxford English Dictionary: "The word is used by W. Harvey, Exercitationes 1651, p. 148, and in the English Anatomical Exercitations 1653, p. 272. It is explained to mean ‘partium super-exorientium additamentum’, ‘the additament of parts budding one out of another’."
- Morange M. La tentative de Nikolai Koltzoff (Koltsov) de lier génétique, embryologie et chimie physique, J. Biosciences. 2011. V. 36. P. 211-214
- Waddington CH (1942). "The epigenotype". Endeavour. 1: 18–20. "For the purpose of a study of inheritance, the relation between phenotypes and genotypes [...] is, from a wider biological point of view, of crucial importance, since it is the kernel of the whole problem of development. Many geneticists have recognized this and attempted to discover the processes involved in the mechanism by which the genes of the genotype bring about phenotypic effects. The first step in such an enterprise is – or rather should be, since it is often omitted by those with undue respect for the powers of reason – to describe what can be seen of the developmental processes. For enquiries of this kind, the word 'phenogenetics' was coined by Haecker [1918, Phänogenetik]. The second and more important part of the task is to discover the causal mechanisms at work and to relate them as far as possible to what experimental embryology has already revealed of the mechanics of development. We might use the name 'epigenetics' for such studies, thus emphasizing their relation to the concepts, so strongly favourable to the classical theory of epigenesis, which have been reached by the experimental embryologists. We certainly need to remember that between genotype and phenotype, and connecting them to each other, there lies a whole complex of developmental processes. It is convenient to have a name for this complex: 'epigenotype' seems suitable."
- See preformationism for historical background. Oxford English Dictionary: "the theory that the germ is brought into existence (by successive accretions), and not merely developed, in the process of reproduction. [...] The opposite theory was formerly known as the 'theory of evolution'; to avoid the ambiguity of this name, it is now spoken of chiefly as the 'theory of preformation', sometimes as that of 'encasement' or 'emboîtement'."
- Waddington CH (2014). The Epigenetics of Birds. Cambridge University Press. ISBN 978-1-107-44047-0.
- Hall BK (January 2004). "In search of evolutionary developmental mechanisms: the 30-year gap between 1944 and 1974". Journal of Experimental Zoology Part B: Molecular and Developmental Evolution. 302 (1): 5–18. doi:10.1002/jez.b.20002. PMID 14760651.
- Alvarez-Buylla ER, Chaos A, Aldana M, Benítez M, Cortes-Poza Y, Espinosa-Soto C, et al. (3 November 2008). "Floral morphogenesis: stochastic explorations of a gene network epigenetic landscape". PLOS ONE. 3 (11): e3626. Bibcode:2008PLoSO...3.3626A. doi:10.1371/journal.pone.0003626. PMC 2572848. PMID 18978941.
- Rabajante JF, Babierra AL (March 2015). "Branching and oscillations in the epigenetic landscape of cell-fate determination". Progress in Biophysics and Molecular Biology. 117 (2–3): 240–249. doi:10.1016/j.pbiomolbio.2015.01.006. PMID 25641423. S2CID 2579314.
- Holliday R (January 1990). "DNA methylation and epigenetic inheritance". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 326 (1235): 329–38. Bibcode:1990RSPTB.326..329H. doi:10.1098/rstb.1990.0015. PMID 1968668.
- Riggs AD, Martienssen RA, Russo VE (1996). Epigenetic mechanisms of gene regulation. Plainview, NY: Cold Spring Harbor Laboratory Press. pp. 1–4. ISBN 978-0-87969-490-6.
- Ledford H (October 2008). "Language: Disputed definitions". Nature. 455 (7216): 1023–8. doi:10.1038/4551023a. PMID 18948925.
- Gottlieb G (1991). "Epigenetic systems view of human development". Developmental Psychology. 27 (1): 33–34. doi:10.1037/0012-1649.27.1.33.
- Gottlieb G (January 2007). "Probabilistic epigenesis" (PDF). Developmental Science. 10 (1): 1–11. doi:10.1111/j.1467-7687.2007.00556.x. PMID 17181692.
- "Probabilistic epigenesis" (PDF).
{{cite web}}: CS1 maint: url-status (link) - Felling RJ, Song H (June 2015). "Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery". Experimental Neurology. Epigenetics in Neurodevelopment and Neurological Diseases. 268: 37–45. doi:10.1016/j.expneurol.2014.09.017. PMC 4375064. PMID 25263580.
- Boeree, C. George, (1997/2006), Personality Theories, Erik Erikson
- Erikson EH (1968). "Chapter 3: The Live Cycle: Epigenesis of Identity". Identity: Youth and Crisis. W.W. Norton and Company. p. 92. ISBN 9780393097863.
- "Epigenetics". Bio-Medicine.org. Retrieved 21 May 2011.
- Yehuda R, Lehrner A (October 2018). "Intergenerational transmission of trauma effects: putative role of epigenetic mechanisms". World Psychiatry. 17 (3): 243–257. doi:10.1002/wps.20568. PMC 6127768. PMID 30192087.
- Lacal I, Ventura R (2018). "Epigenetic Inheritance: Concepts, Mechanisms and Perspectives". Frontiers in Molecular Neuroscience. 11: 292. doi:10.3389/fnmol.2018.00292. PMC 6172332. PMID 30323739.
- "Epigenetic Mechanism - an overview | ScienceDirect Topics". www.sciencedirect.com. Retrieved 3 February 2021.
- Lehrner A, Yehuda R (December 2018). "Cultural trauma and epigenetic inheritance". Development and Psychopathology. 30 (5): 1763–1777. doi:10.1017/S0954579418001153. PMID 30261943. S2CID 52877229.
- Henriques M. "Can the legacy of trauma be passed down the generations?". www.bbc.com. Retrieved 3 February 2021.
- Nestler EJ (March 2016). "Transgenerational Epigenetic Contributions to Stress Responses: Fact or Fiction?". PLOS Biology. 14 (3): e1002426. doi:10.1371/journal.pbio.1002426. PMC 4807775. PMID 27015088.
- Chandler VL (February 2007). "Paramutation: from maize to mice". Cell. 128 (4): 641–5. doi:10.1016/j.cell.2007.02.007. PMID 17320501. S2CID 6928707.
- Zaidi, S. K.; Lian, J. B.; Vanwijnen, A. J.; Stein, J. L.; Stein, G. S. (2017). "Mitotic Gene Bookmarking: An Epigenetic Mechanism for Coordination of Lineage Commitment, Cell Identity and Cell Growth". Advances in Experimental Medicine and Biology. 962: 95–102. doi:10.1007/978-981-10-3233-2_7. ISBN 978-981-10-3231-8. PMC 7233416. PMID 28299653.
- Suter, C. M.; Martin, D. I. (2009). "Paramutation: the tip of an epigenetic iceberg?". Trends in Genetics. 26 (1): 9–14. doi:10.1016/j.tig.2009.11.003. PMC 3137459. PMID 19945764.
- Ferguson-Smith, Anne C. (2011). "Genomic imprinting: the emergence of an epigenetic paradigm". Nature Reviews Genetics. 12 (8): 565–575. doi:10.1038/nrg3032. PMID 21765458. S2CID 23630392.
{{cite journal}}: CS1 maint: url-status (link) - Kovalchuk O, Baulch JE (January 2008). "Epigenetic changes and nontargeted radiation effects--is there a link?". Environmental and Molecular Mutagenesis. 49 (1): 16–25. doi:10.1002/em.20361. PMID 18172877. S2CID 38705208.
- Ilnytskyy Y, Kovalchuk O (September 2011). "Non-targeted radiation effects-an epigenetic connection". Mutation Research. 714 (1–2): 113–25. doi:10.1016/j.mrfmmm.2011.06.014. PMID 21784089.
- Friedl AA, Mazurek B, Seiler DM (2012). "Radiation-induced alterations in histone modification patterns and their potential impact on short-term radiation effects". Frontiers in Oncology. 2: 117. doi:10.3389/fonc.2012.00117. PMC 3445916. PMID 23050241.
- Cuozzo C, Porcellini A, Angrisano T, Morano A, Lee B, Di Pardo A, et al. (July 2007). "DNA damage, homology-directed repair, and DNA methylation". PLOS Genetics. 3 (7): e110. doi:10.1371/journal.pgen.0030110. PMC 1913100. PMID 17616978.
- O'Hagan HM, Mohammad HP, Baylin SB (August 2008). Lee JT (ed.). "Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island". PLOS Genetics. 4 (8): e1000155. doi:10.1371/journal.pgen.1000155. PMC 2491723. PMID 18704159.
- Malanga M, Althaus FR (June 2005). "The role of poly(ADP-ribose) in the DNA damage signaling network" (PDF). Biochemistry and Cell Biology. 83 (3): 354–64. doi:10.1139/o05-038. PMID 15959561.
- Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, et al. (August 2009). "Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler". Proceedings of the National Academy of Sciences of the United States of America. 106 (33): 13770–4. Bibcode:2009PNAS..10613770G. doi:10.1073/pnas.0906920106. PMC 2722505. PMID 19666485.
- Lin JC, Jeong S, Liang G, Takai D, Fatemi M, Tsai YC, et al. (November 2007). "Role of nucleosomal occupancy in the epigenetic silencing of the MLH1 CpG island". Cancer Cell. 12 (5): 432–44. doi:10.1016/j.ccr.2007.10.014. PMC 4657456. PMID 17996647.
- Tabish AM, Poels K, Hoet P, Godderis L (2012). Chiariotti L (ed.). "Epigenetic factors in cancer risk: effect of chemical carcinogens on global DNA methylation pattern in human TK6 cells". PLOS ONE. 7 (4): e34674. Bibcode:2012PLoSO...734674T. doi:10.1371/journal.pone.0034674. PMC 3324488. PMID 22509344.
- Burdge GC, Hoile SP, Uller T, Thomas NA, Gluckman PD, Hanson MA, Lillycrop KA (2011). Imhof A (ed.). "Progressive, transgenerational changes in offspring phenotype and epigenotype following nutritional transition". PLOS ONE. 6 (11): e28282. Bibcode:2011PLoSO...628282B. doi:10.1371/journal.pone.0028282. PMC 3227644. PMID 22140567.
- Fang M, Chen D, Yang CS (January 2007). "Dietary polyphenols may affect DNA methylation". The Journal of Nutrition. 137 (1 Suppl): 223S–228S. doi:10.1093/jn/137.1.223S. PMID 17182830.
- Olaharski AJ, Rine J, Marshall BL, Babiarz J, Zhang L, Verdin E, Smith MT (December 2005). "The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases". PLOS Genetics. 1 (6): e77. doi:10.1371/journal.pgen.0010077. PMC 1315280. PMID 16362078.
- Kikuno N, Shiina H, Urakami S, Kawamoto K, Hirata H, Tanaka Y, et al. (August 2008). "Genistein mediated histone acetylation and demethylation activates tumor suppressor genes in prostate cancer cells". International Journal of Cancer. 123 (3): 552–60. doi:10.1002/ijc.23590. PMID 18431742. S2CID 4704450.
- Djuric Z, Chen G, Doerge DR, Heilbrun LK, Kucuk O (October 2001). "Effect of soy isoflavone supplementation on markers of oxidative stress in men and women". Cancer Letters. 172 (1): 1–6. doi:10.1016/S0304-3835(01)00627-9. PMID 11595123.
- Kropat C, Mueller D, Boettler U, Zimmermann K, Heiss EH, Dirsch VM, et al. (March 2013). "Modulation of Nrf2-dependent gene transcription by bilberry anthocyanins in vivo". Molecular Nutrition & Food Research. 57 (3): 545–50. doi:10.1002/mnfr.201200504. PMID 23349102.
- Verma M, Rogers S, Divi RL, Schully SD, Nelson S, Joseph Su L, et al. (February 2014). "Epigenetic research in cancer epidemiology: trends, opportunities, and challenges". Cancer Epidemiology, Biomarkers & Prevention. 23 (2): 223–33. doi:10.1158/1055-9965.EPI-13-0573. PMC 3925982. PMID 24326628.
- "Studying epigenetics using ChIP".
{{cite web}}: CS1 maint: url-status (link) - "Combined Immunofluorescence, RNA Fluorescent In Situ Hybridization, and DNA Fluorescent In Situ Hybridization to Study Chromatin Changes, Transcriptional Activity, Nuclear Organization, and X-Chromosome Inactivation".
{{cite web}}: CS1 maint: url-status (link) - "Fluorescence In Situ Hybridization (FISH)".
{{cite web}}: CS1 maint: url-status (link) - Hashimoto, Ko; Kokubun, Shoichi; Itoi, Eiji; Roach, Helmtrud I. (2007). "Improved Quantification of DNA Methylation Using Methylation-Sensitive Restriction Enzymes and Real-Time PCR". Epigenetics. 2 (2): 86–91. doi:10.4161/epi.2.2.4203. PMID 17965602. S2CID 26728480.
{{cite journal}}: CS1 maint: url-status (link) - Guerrero-Bosagna C, Skinner MK (May 2012). "Environmentally induced epigenetic transgenerational inheritance of phenotype and disease". Molecular and Cellular Endocrinology. 354 (1–2): 3–8. doi:10.1016/j.mce.2011.10.004. PMC 3312615. PMID 22020198.
- Jablonka E, Lamb MJ, Lachmann M (September 1992). "Evidence, mechanisms and models for the inheritance of acquired characteristics". J. Theor. Biol. 158 (2): 245–68. doi:10.1016/S0022-5193(05)80722-2.
- Ptashne M (April 2007). "On the use of the word 'epigenetic'". Current Biology. 17 (7): R233-6. doi:10.1016/j.cub.2007.02.030. PMID 17407749. S2CID 17490277.
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. (October 2019). "Metabolic regulation of gene expression by histone lactylation". Nature. 574 (7779): 575–580. Bibcode:2019Natur.574..575Z. doi:10.1038/s41586-019-1678-1. PMC 6818755. PMID 31645732.
- Kumar S, Chinnusamy V, Mohapatra T (2018). "Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond". Frontiers in Genetics. 9: 640. doi:10.3389/fgene.2018.00640. PMC 6305559. PMID 30619465.
- Greenberg MV, Bourc'his D (October 2019). "The diverse roles of DNA methylation in mammalian development and disease". Nature Reviews. Molecular Cell Biology. 20 (10): 590–607. doi:10.1038/s41580-019-0159-6. PMID 31399642. S2CID 199512037.
- Spitz F, Furlong EE (September 2012). "Transcription factors: from enhancer binding to developmental control". Nat Rev Genet. 13 (9): 613–26. doi:10.1038/nrg3207. PMID 22868264. S2CID 205485256.
- Duke CG, Kennedy AJ, Gavin CF, Day JJ, Sweatt JD (July 2017). "Experience-dependent epigenomic reorganization in the hippocampus". Learn Mem. 24 (7): 278–288. doi:10.1101/lm.045112.117. PMC 5473107. PMID 28620075.
- Bernstein C (2022). "DNA Methylation and Establishing Memory". Epigenet Insights. 15: 25168657211072499. doi:10.1177/25168657211072499. PMC 8793415. PMID 35098021.
- Rose NR, Klose RJ (December 2014). "Understanding the relationship between DNA methylation and histone lysine methylation". Biochim Biophys Acta. 1839 (12): 1362–72. doi:10.1016/j.bbagrm.2014.02.007. PMC 4316174. PMID 24560929.
- Li Y, Chen X, Lu C (May 2021). "The interplay between DNA and histone methylation: molecular mechanisms and disease implications". EMBO Rep. 22 (5): e51803. doi:10.15252/embr.202051803. PMC 8097341. PMID 33844406.
- Bendandi A, Patelli AS, Diaspro A, Rocchia W (2020). "The role of histone tails in nucleosome stability: An electrostatic perspective". Comput Struct Biotechnol J. 18: 2799–2809. doi:10.1016/j.csbj.2020.09.034. PMC 7575852. PMID 33133421.
- Stewart MD, Li J, Wong J (April 2005). "Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment". Molecular and Cellular Biology. 25 (7): 2525–2538. doi:10.1128/MCB.25.7.2525-2538.2005. PMC 1061631. PMID 15767660.
- Khan FA (2014). Biotechnology in Medical Sciences. United States: CRC Press (imprint of Taylor & Francis Group, an Informa business). p. 239. ISBN 978-1-4822-2368-2.
- Jenuwein T, Laible G, Dorn R, Reuter G (January 1998). "SET domain proteins modulate chromatin domains in eu- and heterochromatin". Cellular and Molecular Life Sciences. 54 (1): 80–93. doi:10.1007/s000180050127. PMID 9487389. S2CID 7769686.
- Slotkin RK, Martienssen R (April 2007). "Transposable elements and the epigenetic regulation of the genome". Nature Reviews. Genetics. 8 (4): 272–85. doi:10.1038/nrg2072. PMID 17363976. S2CID 9719784.
- Li E, Bestor TH, Jaenisch R (June 1992). "Targeted mutation of the DNA methyltransferase gene results in embryonic lethality". Cell. 69 (6): 915–26. doi:10.1016/0092-8674(92)90611-F. PMID 1606615. S2CID 19879601.
- Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA (June 1999). "The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors". Nucleic Acids Research. 27 (11): 2291–8. doi:10.1093/nar/27.11.2291. PMC 148793. PMID 10325416.
- Leonhardt H, Page AW, Weier HU, Bestor TH (November 1992). "A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei" (PDF). Cell. 71 (5): 865–73. doi:10.1016/0092-8674(92)90561-P. PMID 1423634. S2CID 5995820.
- Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF (September 1997). "Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1". Science. 277 (5334): 1996–2000. doi:10.1126/science.277.5334.1996. PMID 9302295.
- Robertson KD, Wolffe AP (October 2000). "DNA methylation in health and disease". Nature Reviews. Genetics. 1 (1): 11–9. doi:10.1038/35049533. PMID 11262868. S2CID 1915808.
- Li E, Beard C, Jaenisch R (November 1993). "Role for DNA methylation in genomic imprinting". Nature. 366 (6453): 362–5. Bibcode:1993Natur.366..362L. doi:10.1038/366362a0. PMID 8247133. S2CID 4311091.
- Viens A, Mechold U, Brouillard F, Gilbert C, Leclerc P, Ogryzko V (July 2006). "Analysis of human histone H2AZ deposition in vivo argues against its direct role in epigenetic templating mechanisms". Molecular and Cellular Biology. 26 (14): 5325–35. doi:10.1128/MCB.00584-06. PMC 1592707. PMID 16809769.
- Ogryzko VV (April 2008). "Erwin Schroedinger, Francis Crick and epigenetic stability". Biology Direct. 3: 15. doi:10.1186/1745-6150-3-15. PMC 2413215. PMID 18419815.
- Nottke A, Colaiácovo MP, Shi Y (March 2009). "Developmental roles of the histone lysine demethylases". Development. 136 (6): 879–89. doi:10.1242/dev.020966. PMC 2692332. PMID 19234061.
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ (March 2009). "Determination of enriched histone modifications in non-genic portions of the human genome". BMC Genomics. 10: 143. doi:10.1186/1471-2164-10-143. PMC 2667539. PMID 19335899.
- Sneppen K, Micheelsen MA, Dodd IB (15 April 2008). "Ultrasensitive gene regulation by positive feedback loops in nucleosome modification". Molecular Systems Biology. 4 (1): 182. doi:10.1038/msb.2008.21. PMC 2387233. PMID 18414483.
- "Epigenetic cell memory". Cmol.nbi.dk. Archived from the original on 30 September 2011. Retrieved 26 July 2012.
- Dodd IB, Micheelsen MA, Sneppen K, Thon G (May 2007). "Theoretical analysis of epigenetic cell memory by nucleosome modification". Cell. 129 (4): 813–22. doi:10.1016/j.cell.2007.02.053. PMID 17512413. S2CID 16091877.
- Morris KL (2008). "Epigenetic Regulation of Gene Expression". RNA and the Regulation of Gene Expression: A Hidden Layer of Complexity. Norfolk, England: Caister Academic Press. ISBN 978-1-904455-25-7.
- Mattick JS, Amaral PP, Dinger ME, Mercer TR, Mehler MF (January 2009). "RNA regulation of epigenetic processes". BioEssays. 31 (1): 51–9. doi:10.1002/bies.080099. PMID 19154003. S2CID 19293469.
- Choi CQ (25 May 2006). "RNA can be hereditary molecule". The Scientist. Archived from the original on 8 February 2007.
- Wang Z, Yao H, Lin S, Zhu X, Shen Z, Lu G, et al. (April 2013). "Transcriptional and epigenetic regulation of human microRNAs". Cancer Letters. 331 (1): 1–10. doi:10.1016/j.canlet.2012.12.006. PMID 23246373.
- "Browse miRBase by species".
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. (February 2005). "Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs". Nature. 433 (7027): 769–73. Bibcode:2005Natur.433..769L. doi:10.1038/nature03315. PMID 15685193. S2CID 4430576.
- Lee D, Shin C (October 2012). "MicroRNA-target interactions: new insights from genome-wide approaches". Annals of the New York Academy of Sciences. 1271 (1): 118–28. Bibcode:2012NYASA1271..118L. doi:10.1111/j.1749-6632.2012.06745.x. PMC 3499661. PMID 23050973.
- Friedman RC, Farh KK, Burge CB, Bartel DP (January 2009). "Most mammalian mRNAs are conserved targets of microRNAs". Genome Research. 19 (1): 92–105. doi:10.1101/gr.082701.108. PMC 2612969. PMID 18955434.
- Goll MG, Bestor TH (2005). "Eukaryotic cytosine methyltransferases". Annual Review of Biochemistry. 74: 481–514. doi:10.1146/annurev.biochem.74.010904.153721. PMID 15952895. S2CID 32123961.
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. (October 2011). "N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO". Nature Chemical Biology. 7 (12): 885–7. doi:10.1038/nchembio.687. PMC 3218240. PMID 22002720.
- "New research links common RNA modification to obesity". Physorg.com. Retrieved 26 July 2012.
- Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T, et al. (August 2013). "Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure". Antimicrobial Agents and Chemotherapy. 57 (8): 3864–74. doi:10.1128/AAC.00263-13. PMC 3719707. PMID 23733475.
- sRNATarBase 2.0 A comprehensive database of bacterial SRNA targets verified by experiments Archived 26 September 2013 at the Wayback Machine
- "Genomics maps for small non-coding RNA's and their targets in microbial genomes". Archived from the original on 8 June 2017. Retrieved 13 August 2013.
- Yool A, Edmunds WJ (1998). "Epigenetic inheritance and prions". Journal of Evolutionary Biology. 11 (2): 241–42. doi:10.1007/s000360050085.
- Cox BS (1965). "[PSI], a cytoplasmic suppressor of super-suppression in yeast". Heredity. 20 (4): 505–21. doi:10.1038/hdy.1965.65.
- Lacroute F (May 1971). "Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast". Journal of Bacteriology. 106 (2): 519–22. doi:10.1128/JB.106.2.519-522.1971. PMC 285125. PMID 5573734.
- Liebman SW, Sherman F (September 1979). "Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast". Journal of Bacteriology. 139 (3): 1068–71. doi:10.1128/JB.139.3.1068-1071.1979. PMC 218059. PMID 225301.
- True HL, Lindquist SL (September 2000). "A yeast prion provides a mechanism for genetic variation and phenotypic diversity". Nature. 407 (6803): 477–83. Bibcode:2000Natur.407..477T. doi:10.1038/35035005. PMID 11028992. S2CID 4411231.
- Shorter J, Lindquist S (June 2005). "Prions as adaptive conduits of memory and inheritance". Nature Reviews. Genetics. 6 (6): 435–50. doi:10.1038/nrg1616. PMID 15931169. S2CID 5575951.
- Giacomelli MG, Hancock AS, Masel J (February 2007). "The conversion of 3' UTRs into coding regions". Molecular Biology and Evolution. 24 (2): 457–64. doi:10.1093/molbev/msl172. PMC 1808353. PMID 17099057.
- Lancaster AK, Bardill JP, True HL, Masel J (February 2010). "The spontaneous appearance rate of the yeast prion [PSI+] and its implications for the evolution of the evolvability properties of the [PSI+] system". Genetics. 184 (2): 393–400. doi:10.1534/genetics.109.110213. PMC 2828720. PMID 19917766.
- Garcia DM, Campbell EA, Jakobson CM, Tsuchiya M, Shaw EA, DiNardo AL, et al. (September 2021). "A prion accelerates proliferation at the expense of lifespan". eLife. 10: e60917. doi:10.7554/eLife.60917. PMC 8455135. PMID 34545808.
- Sapp J (1991). "Concepts of Organization the Leverage of Ciliate Protozoa". A Conceptual History of Modern Embryology. Developmental Biology. Vol. 7. pp. 229–258. doi:10.1007/978-1-4615-6823-0_11. ISBN 978-1-4615-6825-4. PMID 1804215.
- Sapp J (2003). Genesis: the evolution of biology. Oxford: Oxford University Press. ISBN 978-0-19-515619-5.
- Gray RD, Oyama S, Griffiths PE (2003). Cycles of Contingency: Developmental Systems and Evolution (Life and Mind: Philosophical Issues in Biology and Psychology). Cambridge, Massachusetts: The MIT Press. ISBN 978-0-262-65063-2.
- Serizay J, Dong Y, Jänes J, Chesney M, Cerrato C, Ahringer J (20 February 2020). "Tissue-specific profiling reveals distinctive regulatory architectures for ubiquitous, germline and somatic genes". bioRxiv: 2020.02.20.958579. doi:10.1101/2020.02.20.958579. S2CID 212943176.
- Teif VB, Beshnova DA, Vainshtein Y, Marth C, Mallm JP, Höfer T, Rippe K (August 2014). "Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development". Genome Research. 24 (8): 1285–95. doi:10.1101/gr.164418.113. PMC 4120082. PMID 24812327.
- Buschbeck M, Hake SB (May 2017). "Variants of core histones and their roles in cell fate decisions, development and cancer". Nature Reviews. Molecular Cell Biology. 18 (5): 299–314. doi:10.1038/nrm.2016.166. PMID 28144029. S2CID 3307731.
- Jang CW, Shibata Y, Starmer J, Yee D, Magnuson T (July 2015). "Histone H3.3 maintains genome integrity during mammalian development". Genes & Development. 29 (13): 1377–92. doi:10.1101/gad.264150.115. PMC 4511213. PMID 26159997.
- "The 3D genome". www.nature.com. Retrieved 26 September 2021.
- Bernstein C (2022). "DNA Methylation and Establishing Memory". Epigenet Insights. 15: 25168657211072499. doi:10.1177/25168657211072499. PMC 8793415. PMID 35098021.
- Kitamura T, Ogawa SK, Roy DS, Okuyama T, Morrissey MD, Smith LM, Redondo RL, Tonegawa S (April 2017). "Engrams and circuits crucial for systems consolidation of a memory". Science. 356 (6333): 73–78. Bibcode:2017Sci...356...73K. doi:10.1126/science.aam6808. PMC 5493329. PMID 28386011.
- Stott RT, Kritsky O, Tsai LH (2021). "Profiling DNA break sites and transcriptional changes in response to contextual fear learning". PLOS ONE. 16 (7): e0249691. Bibcode:2021PLoSO..1649691S. doi:10.1371/journal.pone.0249691. PMC 8248687. PMID 34197463.
- Lee BH, Shim JY, Moon HC, Kim DW, Kim J, Yook JS, Kim J, Park HY (July 2022). "Real-time visualization of mRNA synthesis during memory formation in live mice". Proc Natl Acad Sci U S A. 119 (27): e2117076119. Bibcode:2022PNAS..11917076L. doi:10.1073/pnas.2117076119. PMC 9271212. PMID 35776545.
- Tischmeyer W, Grimm R (April 1999). "Activation of immediate early genes and memory formation". Cell Mol Life Sci. 55 (4): 564–74. doi:10.1007/s000180050315. PMID 10357227. S2CID 6923522.
- Oliveira AM, Hemstedt TJ, Bading H (July 2012). "Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities". Nat Neurosci. 15 (8): 1111–3. doi:10.1038/nn.3151. PMID 22751036. S2CID 10590208.
- Sun Z, Xu X, He J, Murray A, Sun MA, Wei X, Wang X, McCoig E, Xie E, Jiang X, Li L, Zhu J, Chen J, Morozov A, Pickrell AM, Theus MH, Xie H (August 2019). "EGR1 recruits TET1 to shape the brain methylome during development and upon neuronal activity". Nat Commun. 10 (1): 3892. Bibcode:2019NatCo..10.3892S. doi:10.1038/s41467-019-11905-3. PMC 6715719. PMID 31467272.
- Manzo M, Wirz J, Ambrosi C, Villaseñor R, Roschitzki B, Baubec T (December 2017). "Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands". EMBO J. 36 (23): 3421–3434. doi:10.15252/embj.201797038. PMC 5709737. PMID 29074627.
- Joels G, Lamprecht R (2014). "Fear memory formation can affect a different memory: fear conditioning affects the extinction, but not retrieval, of conditioned taste aversion (CTA) memory". Front Behav Neurosci. 8: 324. doi:10.3389/fnbeh.2014.00324. PMC 4179742. PMID 25324744.
- Moore LD, Le T, Fan G (January 2013). "DNA methylation and its basic function". Neuropsychopharmacology. 38 (1): 23–38. doi:10.1038/npp.2012.112. PMC 3521964. PMID 22781841.
- Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S (January 2016). "DNA methylation changes in plasticity genes accompany the formation and maintenance of memory". Nat Neurosci. 19 (1): 102–10. doi:10.1038/nn.4194. PMID 26656643. S2CID 1173959.
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ (May 2004). "The involvement of the anterior cingulate cortex in remote contextual fear memory". Science. 304 (5672): 881–3. Bibcode:2004Sci...304..881F. doi:10.1126/science.1094804. PMID 15131309. S2CID 15893863.
- Griesemer J, Haber MH, Yamashita G, Gannett L (March 2005). "Critical Notice: Cycles of Contingency – Developmental Systems and Evolution". Biology & Philosophy. 20 (2–3): 517–44. doi:10.1007/s10539-004-0836-4. S2CID 2995306.
- Chapter: "Nervous System Development" in "Epigenetics," by Benedikt Hallgrimsson and Brian Hall
- Costa S, Shaw P (March 2007). "'Open minded' cells: how cells can change fate" (PDF). Trends in Cell Biology. 17 (3): 101–6. doi:10.1016/j.tcb.2006.12.005. PMID 17194589. Archived from the original (PDF) on 15 December 2013.
This might suggest that plant cells do not use or require a cellular memory mechanism and just respond to positional information. However, it has been shown that plants do use cellular memory mechanisms mediated by PcG proteins in several processes, ... (p. 104)
- Cooney CA, Dave AA, Wolff GL (August 2002). "Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring". The Journal of Nutrition. 132 (8 Suppl): 2393S–2400S. doi:10.1093/jn/132.8.2393S. PMID 12163699.
- Waterland RA, Jirtle RL (August 2003). "Transposable elements: targets for early nutritional effects on epigenetic gene regulation". Molecular and Cellular Biology. 23 (15): 5293–300. doi:10.1128/MCB.23.15.5293-5300.2003. PMC 165709. PMID 12861015.
- Dolinoy DC (August 2008). "The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome". Nutrition Reviews. 66 (Suppl 1): S7-11. doi:10.1111/j.1753-4887.2008.00056.x. PMC 2822875. PMID 18673496.
- Schulze KV, Bhatt A, Azamian MS, Sundgren NC, Zapata GE, Hernandez P, et al. (November 2019). "Aberrant DNA methylation as a diagnostic biomarker of diabetic embryopathy". Genetics in Medicine. 21 (11): 2453–2461. doi:10.1038/s41436-019-0516-z. PMID 30992551. S2CID 116880337.
- Fearful Memories Passed Down to Mouse Descendants: Genetic imprint from traumatic experiences carries through at least two generations, By Ewen Callaway and Nature magazine | Sunday, 1 December 2013.
- Mice can 'warn' sons, grandsons of dangers via sperm, by Mariette Le Roux, 12/1/13.
- Francis G (October 2014). "Too much success for recent groundbreaking epigenetic experiments". Genetics. 198 (2): 449–451. doi:10.1534/genetics.114.163998. PMC 4196602. PMID 25316784.
- Dias BG, Ressler KJ (January 2014). "Parental olfactory experience influences behavior and neural structure in subsequent generations". Nature Neuroscience. 17 (1): 89–96. doi:10.1038/nn.3594. PMC 3923835. PMID 24292232. (see comment by Gonzalo Otazu)
- "Epigenetics Paper Raises Questions".
- Hoekstra RF (2000). Evolution: an introduction. Oxford: Oxford University Press. p. 285. ISBN 978-0-19-854968-0.
- Lamb MJ, Jablonka E (2005). Evolution in four dimensions: genetic, epigenetic, behavioral, and symbolic variation in the history of life. Cambridge, Massachusetts: MIT Press. ISBN 978-0-262-10107-3.
- See also Denis Noble: The Music of Life, esp pp. 93–98 and p. 48, where he cites Jablonka & Lamb and Massimo Pigliucci's review of Jablonka and Lamb in Nature 435, 565–566 (2 June 2005)
- Danchin É, Charmantier A, Champagne FA, Mesoudi A, Pujol B, Blanchet S (June 2011). "Beyond DNA: integrating inclusive inheritance into an extended theory of evolution". Nature Reviews. Genetics. 12 (7): 475–86. doi:10.1038/nrg3028. PMID 21681209. S2CID 8837202.
- Maynard Smith J (March 1990). "Models of a dual inheritance system". Journal of Theoretical Biology. 143 (1): 41–53. Bibcode:1990JThBi.143...41M. doi:10.1016/S0022-5193(05)80287-5. PMID 2359317.
- Lynch M (May 2007). "The frailty of adaptive hypotheses for the origins of organismal complexity". Proceedings of the National Academy of Sciences of the United States of America. 104 (Suppl 1): 8597–604. Bibcode:2007PNAS..104.8597L. doi:10.1073/pnas.0702207104. PMC 1876435. PMID 17494740.
- Dickins TE, Rahman Q (August 2012). "The extended evolutionary synthesis and the role of soft inheritance in evolution". Proceedings. Biological Sciences. 279 (1740): 2913–21. doi:10.1098/rspb.2012.0273. PMC 3385474. PMID 22593110.
- Rando OJ, Verstrepen KJ (February 2007). "Timescales of genetic and epigenetic inheritance". Cell. 128 (4): 655–68. doi:10.1016/j.cell.2007.01.023. PMID 17320504. S2CID 17964015.
- Lancaster AK, Masel J (September 2009). "The evolution of reversible switches in the presence of irreversible mimics". Evolution; International Journal of Organic Evolution. 63 (9): 2350–62. doi:10.1111/j.1558-5646.2009.00729.x. PMC 2770902. PMID 19486147.
- van der Graaf A, Wardenaar R, Neumann DA, Taudt A, Shaw RG, Jansen RC, et al. (May 2015). "Rate, spectrum, and evolutionary dynamics of spontaneous epimutations". Proceedings of the National Academy of Sciences of the United States of America. 112 (21): 6676–81. Bibcode:2015PNAS..112.6676V. doi:10.1073/pnas.1424254112. PMC 4450394. PMID 25964364.
- Griswold CK, Masel J (June 2009). "Complex adaptations can drive the evolution of the capacitor [PSI], even with realistic rates of yeast sex". PLOS Genetics. 5 (6): e1000517. doi:10.1371/journal.pgen.1000517. PMC 2686163. PMID 19521499.
- Jablonka E, Raz G (June 2009). "Transgenerational epigenetic inheritance: prevalence, mechanisms, and implications for the study of heredity and evolution" (PDF). The Quarterly Review of Biology. 84 (2): 131–76. CiteSeerX 10.1.1.617.6333. doi:10.1086/598822. PMID 19606595. S2CID 7233550. Archived from the original (PDF) on 15 July 2011. Retrieved 1 November 2017.
- Davies, Hazel (2008). Do Butterflies Bite?: Fascinating Answers to Questions about Butterflies and Moths (Animals Q&A). Rutgers University Press.
- Lewis ZA, Honda S, Khlafallah TK, Jeffress JK, Freitag M, Mohn F, et al. (March 2009). "Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa". Genome Research. 19 (3): 427–37. doi:10.1101/gr.086231.108. PMC 2661801. PMID 19092133.
- Tost J (2008). Epigenetics. Norfolk, England: Caister Academic Press. ISBN 978-1-904455-23-3.
- Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, et al. (January 2013). "Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases". Genome Research. 23 (1): 129–41. doi:10.1101/gr.136739.111. PMC 3530673. PMID 23093720.
- Davis BM, Chao MC, Waldor MK (April 2013). "Entering the era of bacterial epigenomics with single molecule real time DNA sequencing". Current Opinion in Microbiology. 16 (2): 192–8. doi:10.1016/j.mib.2013.01.011. PMC 3646917. PMID 23434113.
- Lluch-Senar M, Luong K, Lloréns-Rico V, Delgado J, Fang G, Spittle K, et al. (2013). Richardson PM (ed.). "Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution". PLOS Genetics. 9 (1): e1003191. doi:10.1371/journal.pgen.1003191. PMC 3536716. PMID 23300489.
- Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, et al. (December 2012). "The methylomes of six bacteria". Nucleic Acids Research. 40 (22): 11450–62. doi:10.1093/nar/gks891. PMC 3526280. PMID 23034806.
- Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, et al. (December 2012). "Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing". Nature Biotechnology. 30 (12): 1232–9. doi:10.1038/nbt.2432. PMC 3879109. PMID 23138224.
- Oliveira PH (August 2021). "Bacterial Epigenomics: Coming of Age". mSystems. 6 (4): e0074721. doi:10.1128/mSystems.00747-21. PMC 8407109. PMID 34402642. S2CID 237149441.
- Casadesús J, Low D (September 2006). "Epigenetic gene regulation in the bacterial world". Microbiology and Molecular Biology Reviews. 70 (3): 830–56. doi:10.1128/MMBR.00016-06. PMC 1594586. PMID 16959970.
- Manso AS, Chai MH, Atack JM, Furi L, De Ste Croix M, Haigh R, et al. (September 2014). "A random six-phase switch regulates pneumococcal virulence via global epigenetic changes". Nature Communications. 5: 5055. Bibcode:2014NatCo...5.5055M. doi:10.1038/ncomms6055. PMC 4190663. PMID 25268848.
- Oliveira PH, Ribis JW, Garrett EM, Trzilova D, Kim A, Sekulovic O, et al. (January 2020). "Epigenomic characterization of Clostridioides difficile finds a conserved DNA methyltransferase that mediates sporulation and pathogenesis". Nature Microbiology. 5 (1): 166–180. doi:10.1038/s41564-019-0613-4. PMC 6925328. PMID 31768029.
- Chahwan R, Wontakal SN, Roa S (March 2011). "The multidimensional nature of epigenetic information and its role in disease". Discovery Medicine. 11 (58): 233–43. PMID 21447282.
- Beil L (Winter 2008). "Medicine's New Epicenter? Epigenetics: New field of epigenetics may hold the secret to flipping cancer's "off" switch". CURE (Cancer Updates, Research and Education). Archived from the original on 29 May 2009.
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. (July 2005). "Epigenetic differences arise during the lifetime of monozygotic twins". Proceedings of the National Academy of Sciences of the United States of America. 102 (30): 10604–9. Bibcode:2005PNAS..10210604F. doi:10.1073/pnas.0500398102. PMC 1174919. PMID 16009939.
- Kaminsky ZA, Tang T, Wang SC, Ptak C, Oh GH, Wong AH, et al. (February 2009). "DNA methylation profiles in monozygotic and dizygotic twins". Nature Genetics. 41 (2): 240–5. doi:10.1038/ng.286. PMID 19151718. S2CID 12688031.
- O'Connor A (11 March 2008). "The Claim: Identical Twins Have Identical DNA". New York Times. Retrieved 2 May 2010.
- Ballestar E (August 2010). "Epigenetics lessons from twins: prospects for autoimmune disease". Clinical Reviews in Allergy & Immunology. 39 (1): 30–41. doi:10.1007/s12016-009-8168-4. PMID 19653134. S2CID 25040280.
- Wallace RG, Twomey LC, Custaud MA, Moyna N, Cummins PM, Mangone M, Murphy RP (2016). "Potential Diagnostic and Prognostic Biomarkers of Epigenetic Drift within the Cardiovascular Compartment". BioMed Research International. 2016: 2465763. doi:10.1155/2016/2465763. PMC 4749768. PMID 26942189.
- Online Mendelian Inheritance in Man (OMIM): 105830
- Wood AJ, Oakey RJ (November 2006). "Genomic imprinting in mammals: emerging themes and established theories". PLOS Genetics. 2 (11): e147. doi:10.1371/journal.pgen.0020147. PMC 1657038. PMID 17121465.
- Knoll JH, Nicholls RD, Magenis RE, Graham JM, Lalande M, Latt SA (February 1989). "Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion". American Journal of Medical Genetics. 32 (2): 285–90. doi:10.1002/ajmg.1320320235. PMID 2564739.
- Alessio N, Riccitiello F, Squillaro T, Capasso S, Del Gaudio S, Di Bernardo G, et al. (March 2018). "Neural stem cells from a mouse model of Rett syndrome are prone to senescence, show reduced capacity to cope with genotoxic stress, and are impaired in the differentiation process". Experimental & Molecular Medicine. 50 (3): 1. doi:10.1038/s12276-017-0005-x. PMC 6118406. PMID 29563495.
- A person's paternal grandson is the son of a son of that person; a maternal grandson is the son of a daughter.
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjöström M, Golding J (February 2006). "Sex-specific, male-line transgenerational responses in humans". European Journal of Human Genetics. 14 (2): 159–66. doi:10.1038/sj.ejhg.5201538. PMID 16391557. Robert Winston refers to this study in a lecture Archived 23 May 2007 at the Wayback Machine; see also discussion at Leeds University, here
- "NOVA | Transcripts | Ghost in Your Genes". PBS. 16 October 2007. Retrieved 26 July 2012.
- Basu Mallik S, Jayashree BS, Shenoy RR (May 2018). "Epigenetic modulation of macrophage polarization- perspectives in diabetic wounds". Journal of Diabetes and Its Complications. 32 (5): 524–530. doi:10.1016/j.jdiacomp.2018.01.015. PMID 29530315.
- Anderson SJ, Feye KM, Schmidt-McCormack GR, Malovic E, Mlynarczyk GS, Izbicki P, et al. (May 2016). "Off-Target drug effects resulting in altered gene expression events with epigenetic and "Quasi-Epigenetic" origins". Pharmacological Research. 107: 229–233. doi:10.1016/j.phrs.2016.03.028. PMID 27025785.
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. (July 2003). "Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene". Science. 301 (5631): 386–9. Bibcode:2003Sci...301..386C. doi:10.1126/science.1083968. PMID 12869766. S2CID 146500484.
- Alavian-Ghavanini A, Rüegg J (January 2018). "Understanding Epigenetic Effects of Endocrine Disrupting Chemicals: From Mechanisms to Novel Test Methods". Basic & Clinical Pharmacology & Toxicology. 122 (1): 38–45. doi:10.1111/bcpt.12878. PMID 28842957.
- Coplan J, Chanatry ST, Rosenblum LA (2017). "Persistence of Early-Life Stress on the Epigenome: Nonhuman Primate Observations☆". Reference Module in Neuroscience and Biobehavioral Psychology. doi:10.1016/B978-0-12-809324-5.02862-5. ISBN 9780128093245.
- Plomin R, DeFries JC, Knopik VS, Neiderhiser JM (2017). Behavioral Genetics (Seventh ed.). Worth Publishers. pp. 152–153. ISBN 978-1-4292-4215-8.
- Heard E, Martienssen RA (March 2014). "Transgenerational epigenetic inheritance: myths and mechanisms". Cell. 157 (1): 95–109. doi:10.1016/j.cell.2014.02.045. PMC 4020004. PMID 24679529.
- Robison AJ, Nestler EJ (October 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews. Neuroscience. 12 (11): 623–37. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
- Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues in Clinical Neuroscience. 15 (4): 431–43. doi:10.31887/DCNS.2013.15.4/enestler. PMC 3898681. PMID 24459410.
- Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". The American Journal of Drug and Alcohol Abuse. 40 (6): 428–37. doi:10.3109/00952990.2014.933840. PMID 25083822. S2CID 19157711.
Conclusions
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure. The formation of ΔFosB in multiple brain regions, and the molecular pathway leading to the formation of AP-1 complexes is well understood. The establishment of a functional purpose for ΔFosB has allowed further determination as to some of the key aspects of its molecular cascades, involving effectors such as GluR2 (87,88), Cdk5 (93) and NFkB (100). Moreover, many of these molecular changes identified are now directly linked to the structural, physiological and behavioral changes observed following chronic drug exposure (60,95,97,102). New frontiers of research investigating the molecular roles of ΔFosB have been opened by epigenetic studies, and recent advances have illustrated the role of ΔFosB acting on DNA and histones, truly as a ‘‘molecular switch’’ (34). As a consequence of our improved understanding of ΔFosB in addiction, it is possible to evaluate the addictive potential of current medications (119), as well as use it as a biomarker for assessing the efficacy of therapeutic interventions (121,122,124). Some of these proposed interventions have limitations (125) or are in their infancy (75). However, it is hoped that some of these preliminary findings may lead to innovative treatments, which are much needed in addiction. - Biliński P, Wojtyła A, Kapka-Skrzypczak L, Chwedorowicz R, Cyranka M, Studziński T (2012). "Epigenetic regulation in drug addiction". Annals of Agricultural and Environmental Medicine. 19 (3): 491–6. PMID 23020045.
For these reasons, ΔFosB is considered a primary and causative transcription factor in creating new neural connections in the reward centre, prefrontal cortex, and other regions of the limbic system. This is reflected in the increased, stable and long-lasting level of sensitivity to cocaine and other drugs, and tendency to relapse even after long periods of abstinence. These newly constructed networks function very efficiently via new pathways as soon as drugs of abuse are further taken ... In this way, the induction of CDK5 gene expression occurs together with suppression of the G9A gene coding for dimethyltransferase acting on the histone H3. A feedback mechanism can be observed in the regulation of these 2 crucial factors that determine the adaptive epigenetic response to cocaine. This depends on ΔFosB inhibiting G9a gene expression, i.e. H3K9me2 synthesis which in turn inhibits transcription factors for ΔFosB. For this reason, the observed hyper-expression of G9a, which ensures high levels of the dimethylated form of histone H3, eliminates the neuronal structural and plasticity effects caused by cocaine by means of this feedback which blocks ΔFosB transcription
- Vassoler FM, Sadri-Vakili G (April 2014). "Mechanisms of transgenerational inheritance of addictive-like behaviors". Neuroscience. 264: 198–206. doi:10.1016/j.neuroscience.2013.07.064. PMC 3872494. PMID 23920159.
- Yuan TF, Li A, Sun X, Ouyang H, Campos C, Rocha NB, et al. (November 2016). "Transgenerational Inheritance of Paternal Neurobehavioral Phenotypes: Stress, Addiction, Ageing and Metabolism". Molecular Neurobiology. 53 (9): 6367–6376. doi:10.1007/s12035-015-9526-2. hdl:10400.22/7331. PMID 26572641. S2CID 25694221.
- Short AK, Fennell KA, Perreau VM, Fox A, O'Bryan MK, Kim JH, et al. (June 2016). "Elevated paternal glucocorticoid exposure alters the small noncoding RNA profile in sperm and modifies anxiety and depressive phenotypes in the offspring". Translational Psychiatry. 6 (6): e837. doi:10.1038/tp.2016.109. PMC 4931607. PMID 27300263.
- Chahwan R, Wontakal SN, Roa S (October 2010). "Crosstalk between genetic and epigenetic information through cytosine deamination". Trends in Genetics. 26 (10): 443–8. doi:10.1016/j.tig.2010.07.005. PMID 20800313.
- Badal S, Her YF, Maher LJ (September 2015). "Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells". The Journal of Biological Chemistry. 290 (36): 22287–97. doi:10.1074/jbc.M115.671222. PMC 4571980. PMID 26205818.
- Mezentseva NV, Yang J, Kaur K, Iaffaldano G, Rémond MC, Eisenberg CA, Eisenberg LM (February 2013). "The histone methyltransferase inhibitor BIX01294 enhances the cardiac potential of bone marrow cells". Stem Cells and Development. 22 (4): 654–67. doi:10.1089/scd.2012.0181. PMC 3564468. PMID 22994322.
- Yang J, Kaur K, Ong LL, Eisenberg CA, Eisenberg LM (2015). "Inhibition of G9a Histone Methyltransferase Converts Bone Marrow Mesenchymal Stem Cells to Cardiac Competent Progenitors". Stem Cells International. 2015: 270428. doi:10.1155/2015/270428. PMC 4454756. PMID 26089912.
- "Epigenetics: It doesn't mean what quacks think it means". Science-Based Medicine.
Further reading
- Haque FN, Gottesman II, Wong AH (May 2009). "Not really identical: epigenetic differences in monozygotic twins and implications for twin studies in psychiatry". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 151C (2): 136–41. doi:10.1002/ajmg.c.30206. PMID 19378334. S2CID 205327825.
External links
- "Epigenetics & Inheritance". learn.genetics.utah.edu. Retrieved 17 April 2019.
- The Human Epigenome Project (HEP)
- The Epigenome Network of Excellence (NoE)
- Canadian Epigenetics, Environment and Health Research Consortium (CEEHRC)
- The Epigenome Network of Excellence (NoE) – public international site
- "DNA Is Not Destiny" – Discover magazine cover story
- "The Ghost In Your Genes", Horizon (2005), BBC
- Epigenetics article at Hopkins Medicine
- Towards a global map of epigenetic variation
| |
| Key components | |
| Fields |
|
| Archaeogenetics of |
|
| Related topics |
|
| Lists |
|
| |
| |||||||
| Overview |
| ||||||
| Engineering |
| ||||||
| |||||||