Limb–girdle muscular dystrophy
Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics.[7] It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles.[8] LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.[3][9]
| Limb–girdle muscular dystrophy | |
|---|---|
| Other names | Erb's muscular dystrophy[1] |
 | |
| Protein MYOT (also known as TTID one of the many genes whose mutations are responsible for this condition) | |
| Specialty | Neurology, neuromuscular medicine |
| Symptoms | Pelvic muscle weakness[2] |
| Duration | Lifelong |
| Types | 32 types[3] |
| Causes | Genetic mutations |
| Diagnostic method | Genetic testing, and possibly muscle biopsy[4] |
| Differential diagnosis | Muscular dystrophies: Duchenne, Becker, facioscapulohumeral, Emery-Dreifuss; Pompe disease; congenital myasthenic syndrome; motor neuropathy |
| Treatment | Occupational, speech and physical therapy[5] |
| Frequency | 2.27-10 per 100,000[6] |
Signs and symptoms
By definition, all limb girdle muscular dystrophies (LGMD) cause progressive proximal weakness,[3] meaning weakness of the muscles on or close to the torso that worsens over time. Explicitly, LGMD preferentially affects muscles of the hip girdle, thigh, shoulder girdle, and/or upper arm.[8][6] The muscle weakness is generally symmetric.[10] Usually, the hip girdle is the first area to exhibit weakness,[2] manifesting as difficulty walking, going up and/or down stairs, rising from a chair, bending at the waist, or squatting. Because of these difficulties, falling can occur frequently. Weakness of the shoulder girdle can make lifting objects, or even elevating the arms, difficult or impossible. Rate of progression varies between patients. Eventually, the abilities to run and walk can deteriorate.[2][4] The disease commonly leads to dependence on a wheelchair within years of symptom onset, although some patients maintain mobility.[11][2] Eventually the disease can affect other muscles such as the ones located in the face.
By definition, LGMDs primarily affect skeletal muscles,[3] although cardiac muscle can be affected to a lesser degree in select subtypes, which can cause palpitations
There can be significant variability in disease features and severity between LGMD subtypes, and even within any given LGMD subtype.[10] Additional possible presentations include:
- Pseudohypertrophy[2]
- Muscle hypertrophy[12]
- Respiratory muscle problems[12]
- Low back discomfort[2]
- Distal muscle problems[12]
- Winged scapula
In most cases, pain is not present with LGMD, and mental function is not affected. LGMD can begin in childhood, adolescence, young adulthood or even later, the age of onset is usually between 10 and 30. Both genders are affected equally. When limb-girdle muscular dystrophy begins in childhood, the progression appears to be faster and the disease more disabling. When the disorder begins in adolescence or adulthood the disease is generally not as severe and progresses more slowly. There is no sensory neuropathy or autonomic or visceral dysfunction at presentation.
Genetics

LGMD is a genetic and heritable disorder, due to one of many genetic mutations of proteins involved in muscle function. All currently identified LGMDs have an inheritance pattern that is dominant or recessive, although the definition of LGMD allows for diseases with more complicated inheritance patterns to be classified as LGMD. Examples of proteins affected in LGMD are α, β, γ and δ sarcoglycans.
Diagnosis
The diagnosis of limb–girdle muscular dystrophy can be done via muscle biopsy, which will show the presence of muscular dystrophy, and genetic testing is used to determine which type of muscular dystrophy a patient has. Immunohistochemical dystrophin tests can indicate a decrease in dystrophin detected in sarcoglycanopathies. In terms of sarcoglycan deficiency there can be variance (if α-sarcoglycan and γ-sarcoglycan are not present then there's a mutation in LGMD2D).[4]
The 2014 Evidence-based guideline summary: Diagnosis and treatment of limb–girdle and distal dystrophies indicates that individuals suspected of having the inherited disorder should have genetic testing. Other tests/analysis are:[4][5]
- High CK levels(x10-150 times normal)
- MRI can indicate different types of LGMD.
- EMG can confirm the myopathic characteristic of the disease.
- Electrocardiography (cardiac arrhythmias in LGMD1B can occur)
Types
The "LGMD D" family is autosomal dominant, and the "LGMD R" family is autosomal recessive.[3] Limb–girdle muscular dystrophy is explained in terms of gene, locus, OMIM and type as follows:
| Name[3] | Inheritance | Old Name[11] | OMIM | Gene | Gene also implicated in: | Notes |
|---|---|---|---|---|---|---|
| LGMD D1 DNAJB6-related | Autosomal dominant | LGMD1D | 603511 | DNAJB6 | ||
| LGMD D2 TNP03-related | LGMD1F | 608423 | TNPO3 | |||
| LGMD D3 HNRNPDL-related | LGMD1G | 609115 | HNRPDL | |||
| LGMD D4 calpain3-related | LGMD1I | 618129 | CAPN3 | LGMD R1 | Also referred to as "autosomal dominant calpainopathy." | |
| LGMD D5 collagen 6-related | Bethlem myopathy dominant | 158810 | COL6A1, COL6A2, COL6A3 | Congenital muscular dystrophy, LGMD R22 | ||
| LGMD R1 calpain3-related | Autosomal recessive | LGMD2A | 253600 | CAPN3 | LGMD D4 | Also referred to as "autosomal recessive calpainopathy."[13] |
| LGMD R2 dysferlin-related | LGMD2B | 253601 | DYSF | Miyoshi myopathy type 1 (MMD1 - 254130).[14] | A dysferlinopathy | |
| LGMD R3 α-sarcoglycan-related | LGMD2D | 608099 | SGCA | sarcoglycanopathies | ||
| LGMD R4 β -sarcoglycan-related | LGMD2E | 604286 | SGCB | |||
| LGMD R5 γ -sarcoglycan-related | LGMD2C | 253700 | SGCG | |||
| LGMD R6 δ-sarcoglycan-related | LGMD2F | 601287 | SGCD | |||
| LGMD R7 telethonin-related | LGMD2G | 601954 | TCAP | |||
| LGMD R8 TRIM 32-related | LGMD2H | 254110 | TRIM32 | |||
| LGMD R9 FKRP-related | LGMD2I | 607155 | FKRP | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R10 titin-related | LGMD2J | 608807 | TTN | Congenital myopathy | ||
| LGMD R11 POMT1-related | LGMD2K | 609308 | POMT1 | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R12 anoctamin5-related | LGMD2L | 611307 | ANO5 | Miyoshi myopathy type 3 (MMD3 - 613319) | ||
| LGMD R13 Fukutin-related | LGMD2M | 611588 | FKTN | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R14 POMT2-related | LGMD2N | 607439 | POMT2 | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R15 POMGnT1-related | LGMD2O | 606822 | POMGNT1 | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R16 α-dystroglycan-related | LGMD2P | 613818 | DAG1 | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R17 plectin-related | LGMD2Q | 613723 | PLEC1 | |||
| LGMD R18 TRAPPC11-related | LGMD2S | 615356 | TRAPPC11 | |||
| LGMD R19 GMPPB-related | LGMD2T | 615352 | GMPPB | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R20 ISPD-related | LGMD2U | 616052 | ISPD | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R21 POGLUT1-related | LGMD2Z | 617232 | POGLUT1 | |||
| LGMD R22 collagen 6-related | Bethlem myopathy recessive | COL6A1, COL6A2, COL6A3 | Congenital muscular dystrophy, LGMD D5 | |||
| LGMD R23 laminin α2-related | Laminin α2-related muscular dystrophy | 156225 | LAMA2 | Congenital muscular dystrophy | ||
| LGMD R24 POMGNT2-related | POMGNT2-related muscular dystrophy | 618135 | POMGNT2 | Congenital muscular dystrophy | An α-dystroglycanopathy | |
| LGMD R25[7] | LGMD2X | POPDC1 | ||||
| LGMD R26[15] | POPDC3 | |||||
| LGMD R27[16] | JAG2 | |||||
| LGMD R(number pending)[7] | PYROXD1 |
LGMD criteria
For a disease entity to be classified as an LGMD, the following criteria must be met:[3]
- genetic, with an identifiable inheritance pattern such as autosomal dominant, autosomal recessive, digenic, or polygenic.
- relatively selective to skeletal muscle
- predominantly proximal muscle involvement
- independent walking is achieved at one point in life
- elevated serum creatine kinase
- muscle fiber loss
- dystrophic changes in muscle histology
- degenerative changes on medical imaging
- end-stage pathology seen in the most affected muscles
- described in at least two unrelated families
Differential
Many diseases can manifest similarly to LGMD.[6] Dystrophinopathies, including Duchenne muscular dystrophy, Becker muscular dystrophy, and manifesting dystrophinopathy in female carriers, can present similarly to LGMD.[6] Facioscapulohumeral muscular dystrophy can appear similarly, especially when it spares the facial muscles.[6] Also in the differential are Emery-Dreifuss muscular dystrophies, Pompe disease, later-onset congenital myasthenic syndromes, and proximal-predominant hereditary motor neuropathies.[6]
Treatment

There are few studies corroborating the effectiveness of exercise for limb–girdle muscular dystrophy. However studies have shown that exercise can, in fact, damage muscles permanently due to intense muscle contraction.[17] Physical therapy may be required to maintain as much muscle strength and joint flexibility as possible. Calipers may be used to maintain mobility and quality of life. Careful attention to lung and heart health is required, corticosteroids in LGMD 2C-F individuals, shows some improvement.[12] Additionally individuals can follow management that follows:[5]
- Occupational therapy
- Respiratory therapy
- Speech therapy
- Neutralizing antibody to myostatin should not be pursued
The sarcoglycanopathies could be possibly amenable to gene therapy.[18]
Prognosis
In terms of the prognosis of limb–girdle muscular dystrophy in its mildest form, affected individuals have near-normal muscle strength and function. LGMD isn't typically a fatal disease, though it may eventually weaken the heart and respiratory muscles, leading to illness or death due to secondary disorders.
Epidemiology
The minimum prevalence of limb–girdle muscular dystrophy, as a group, likely ranges from 2.27-10 per 100,000 (1:44,000 to 1:10,000).[6] LGMD is the fourth most common muscular dystrophy, after the dystrophinopathies, myotonic dystrophies, and facioscapulohumeral muscular dystrophy.[19] The prevalence of individual LGMDs, as studied in the United States, in descending order, are those due to mutation of 1) calpain, 2) dysferlin, 3) collagen VI, 4) sarcoglycans, 5) anoctamin 5, and 6) fukutin-related protein.[6] It is difficult to calculate the worldwide prevalence of even the most common LGMD types, due to the founder effect causing varying prevalence by region.[7] The less common types are very rare, often only described is limited regions of the world.[7]
History
The term 'limb girdle muscular dystrophy' was published in 1954, describing a group of heterogeneous conditions that clinicians noticed to be distinct from Duchenne muscular dystrophy, facioscapulohumeral muscular dystrophy, and myotonic dystrophy.[3] The genetics of LGMDs began to be understood in the late 1900s, which led the European Neuromuscular Centre (ENMC) to establish a consensus on classification of LGMDs in 1995.[3] The classification scheme at that time denoted autosomal dominant LGMDs as 'LGMD1' and autosomal recessive LGMDs as 'LGMD2.'[3] A letter was appended to the names of LGMDs according to the order of discovery of the causal genetic mutation.[3] As LGMD2Z was established, the question arose of what letter to assign the next discovered LGMD2.[3] With this issue, among other motives, the ENMC established a new consensus on the classification and definition of LGMD in 2017.[3] With the new definition, several diseases were removed from the LGMD category:
| Current name | Old Name | OMIM | Gene | Locus | Reason for exclusion |
|---|---|---|---|---|---|
| Myofibrillar myopathy | LGMD1A | 159000 | TTID | Distal weakness | |
| Emery–Dreifuss muscular dystrophy (EDMD) | LGMD1B | 159001 | LMNA | EDMD phenotype and significant cardiac involvement | |
| Rippling muscle disease | LGMD1C | 607801 | CAV3 | Mainly characterized by muscle rippling and pain | |
| Myofibrillar myopathy | LGMD1E | 601419 | DES | Distal weakness and significant cardiac involvement | |
| Not confirmed | LGMD1H | 613530 | 3p25.1–p23 | Still not established as a disease |
Research
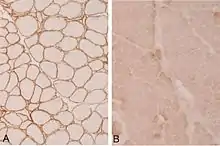
There is a variety of research under way targeted at various forms of limb–girdle muscular dystrophy. Among the treatments thought to hold promise is gene therapy, which is the delivery of genetic material, often a copy of a healthy gene, into cells.[20]
According to a review by Bengtsson et al. some success with AAV-mediated gene therapies (for different disorders) have increased interest in researchers, with CRISPR/Cas9 and exon-skipping helping these therapeutic goals along. Limb–girdle muscular dystrophies have many different types which are due to different gene mutations. LGMD2D is caused by a mutation in the α-sarcoglycan gene. Future treatment could be had by gene therapy through recombinant adeno-associated vectors.[21]
According to a review by Straub, et al., there are several research issues that need to be addressed: the rareness of the disease, poor understanding of the mechanism of LGMD R, and absence of patient cohorts, all contributing to lack of biomarkers for LGMD. The review goes on to state that animal models for LGMD R have been used to analyze therapeutic medications. Also, although prednisone has been used and has had positive effects on affected LGMD2 individuals, there is still no evidence of its effectiveness in trials that are placebo-controlled.[22]
See also
References
- Newfoundland, FRCP William Pryse-Phillips MD, FRCP(C) Faculty of Medicine Health Sciences Centre Memorial University of Newfoundland St John's (2009-05-06). Companion to Clinical Neurology. Oxford University Press, USA. p. 579. ISBN 9780199710041.
- MedlinePlus Encyclopedia: Limb-girdle muscular dystrophies
- Straub, V; Murphy, A; Udd, B; LGMD workshop study, group. (August 2018). "229th ENMC international workshop: Limb girdle muscular dystrophies - Nomenclature and reformed classification Naarden, the Netherlands, 17-19 March 2017". Neuromuscular Disorders. 28 (8): 702–710. doi:10.1016/j.nmd.2018.05.007. PMID 30055862. S2CID 51865029.
- "Limb-Girdle Muscular Dystrophy: Practice Essentials, Background, Pathophysiology". August 2018.
{{cite journal}}: Cite journal requires|journal=(help) - Narayanaswami, Pushpa; Weiss, Michael; Selcen, Duygu; David, William; Raynor, Elizabeth; Carter, Gregory; Wicklund, Matthew; Barohn, Richard J.; Ensrud, Erik (2014-10-14). "Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies". Neurology. 83 (16): 1453–1463. doi:10.1212/WNL.0000000000000892. ISSN 0028-3878. PMC 4206155. PMID 25313375.
- Wicklund, MP (December 2019). "The Limb-Girdle Muscular Dystrophies". Continuum (Minneapolis, Minn.). 25 (6): 1599–1618. doi:10.1212/CON.0000000000000809. PMID 31794462. S2CID 208531741.
- Barton, ER; Pacak, CA; Stoppel, WL; Kang, PB (29 July 2020). "The ties that bind: functional clusters in limb-girdle muscular dystrophy". Skeletal Muscle. 10 (1): 22. doi:10.1186/s13395-020-00240-7. PMC 7389686. PMID 32727611.
- "Limb-girdle muscular dystrophy".
- Pozsgai, E; Griffin, D; Potter, R; Sahenk, Z; Lehman, K; Rodino-Klapac, LR; Mendell, JR (October 2021). "Unmet needs and evolving treatment for limb girdle muscular dystrophies". Neurodegenerative Disease Management. 11 (5): 411–429. doi:10.2217/nmt-2020-0066. PMID 34472379. S2CID 237389009.
- Murphy, AP; Straub, V (22 July 2015). "The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies". Journal of Neuromuscular Diseases. 2 (s2): S7–S19. doi:10.3233/JND-150105. PMC 5271430. PMID 27858764.
- Pegoraro, Elena; Hoffman, Eric P. (1993-01-01). "Limb-Girdle Muscular Dystrophy Overview – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY". In Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Limb-Girdle Muscular Dystrophy Overview. Seattle (WA): University of Washington, Seattle. PMID 20301582.update 2012
- "Limb-girdle Muscular Dystrophy | Doctor".
- Lasa-Elgarresta, J; Mosqueira-Martín, L; Naldaiz-Gastesi, N; Sáenz, A; López de Munain, A; Vallejo-Illarramendi, A (13 September 2019). "Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations". International Journal of Molecular Sciences. 20 (18): 4548. doi:10.3390/ijms20184548. PMC 6770289. PMID 31540302.
- Aoki, Masashi (March 5, 2015). "Dysferlinopathy". GeneReviews. PMID 20301480.
- Benarroch, Louise; Bonne, Gisèle; Rivier, François; Hamroun, Dalil (December 2020). "The 2021 version of the gene table of neuromuscular disorders (nuclear genome)". Neuromuscular Disorders. 30 (12): 1008–1048. doi:10.1016/j.nmd.2020.11.009. PMID 33257164. S2CID 227123684.
- Coppens, S; Barnard, AM; Puusepp, S; Pajusalu, S; Õunap, K; Vargas-Franco, D; Bruels, CC; Donkervoort, S; Pais, L; Chao, KR; Goodrich, JK; England, EM; Weisburd, B; Ganesh, VS; Gudmundsson, S; O'Donnell-Luria, A; Nigul, M; Ilves, P; Mohassel, P; Siddique, T; Milone, M; Nicolau, S; Maroofian, R; Houlden, H; Hanna, MG; Quinlivan, R; Beiraghi Toosi, M; Ghayoor Karimiani, E; Costagliola, S; Deconinck, N; Kadhim, H; Macke, E; Lanpher, BC; Klee, EW; Łusakowska, A; Kostera-Pruszczyk, A; Hahn, A; Schrank, B; Nishino, I; Ogasawara, M; El Sherif, R; Stojkovic, T; Nelson, I; Bonne, G; Cohen, E; Boland-Augé, A; Deleuze, JF; Meng, Y; Töpf, A; Vilain, C; Pacak, CA; Rivera-Zengotita, ML; Bönnemann, CG; Straub, V; Handford, PA; Draper, I; Walter, GA; Kang, PB (6 May 2021). "A form of muscular dystrophy associated with pathogenic variants in JAG2". American Journal of Human Genetics. 108 (5): 840–856. doi:10.1016/j.ajhg.2021.03.020. PMC 8206160. PMID 33861953.
- Siciliano G, Simoncini C, Giannotti S, Zampa V, Angelini C, Ricci G (2015). "Muscle exercise in limb girdle muscular dystrophies: pitfall and advantages". Acta Myologica. 34 (1): 3–8. PMC 4478773. PMID 26155063.
- Reference, Genetics Home. "limb-girdle muscular dystrophy". Genetics Home Reference. Retrieved 2016-04-22.
- Bockhorst, J; Wicklund, M (August 2020). "Limb Girdle Muscular Dystrophies". Neurologic Clinics. 38 (3): 493–504. doi:10.1016/j.ncl.2020.03.009. PMID 32703463. S2CID 220730696.
- Reference, Genetics Home. "How does gene therapy work?". Genetics Home Reference. Retrieved 2016-04-23.
- Bengtsson, Niclas E.; Seto, Jane T.; Hall, John K.; Chamberlain, Jeffrey S.; Odom, Guy L. (2016-04-15). "Progress and prospects of gene therapy clinical trials for the muscular dystrophies". Human Molecular Genetics. 25 (R1): R9–R17. doi:10.1093/hmg/ddv420. ISSN 0964-6906. PMC 4802376. PMID 26450518.
- Straub, Volker; Bertoli, Marta (2016-02-01). "Where do we stand in trial readiness for autosomal recessive limb girdle muscular dystrophies?". Neuromuscular Disorders. 26 (2): 111–125. doi:10.1016/j.nmd.2015.11.012. PMID 26810373. S2CID 23787096. – via ScienceDirect (Subscription may be required or content may be available in libraries.)
Further reading
- Cotta, Ana; Carvalho, Elmano; da-Cunha-Júnior, Antonio Lopes; Paim, Júlia Filardi; Navarro, Monica M.; Valicek, Jaquelin; Menezes, Miriam Melo; Nunes, Simone Vilela; Xavier Neto, Rafael (2014). "Common recessive limb girdle muscular dystrophies differential diagnosis: why and how?". Arquivos de Neuro-Psiquiatria. 72 (9): 721–734. doi:10.1590/0004-282X20140110. ISSN 0004-282X. PMID 25252238.
- Liu, Jian; Harper, Scott Q. (2012-08-01). "RNAi-based Gene Therapy for Dominant Limb Girdle Muscular Dystrophies". Current Gene Therapy. 12 (4): 307–314. doi:10.2174/156652312802083585. ISSN 1566-5232. PMC 4120526. PMID 22856606.
- ANGELINI, CORRADO; TASCA, ELISABETTA; NASCIMBENI, ANNA CHIARA; FANIN, MARINA (2014-12-01). "Muscle fatigue, nNOS and muscle fiber atrophy in limb girdle muscular dystrophy". Acta Myologica. 33 (3): 119–126. ISSN 1128-2460. PMC 4369848. PMID 25873780.
External links
| Types | |
|---|---|
| National/International Organizations |
|
| National/International Events |
|
| Clinical trials | |