Progestogen ester
A progestogen ester is an ester of a progestogen or progestin (a synthetic progestogen). The prototypical progestogen is progesterone, an endogenous sex hormone. Esterification is frequently employed to improve the pharmacokinetics of steroids, including oral bioavailability, lipophilicity, and elimination half-life.[1] In addition, with intramuscular injection, steroid esters are often absorbed more slowly into the body, allowing for less frequent administration.[1] Many (though not all) steroid esters function as prodrugs.
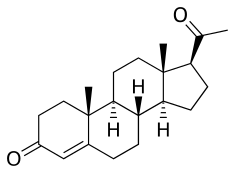
Esterification is particularly salient in the case of progesterone because progesterone itself shows very poor oral pharmacokinetics and is thus ineffective when taken orally.[2][3] Unmodified, it has an elimination half-life of only 5 minutes, and is almost completely inactivated by the liver during first-pass metabolism.[3] Micronization, however, has allowed for progesterone to be effective orally, although oral micronized progesterone was not developed until recent years.[2]
Examples of important progestogen esters include the 17α-hydroxyprogesterone derivatives medroxyprogesterone acetate, megestrol acetate, cyproterone acetate, and hydroxyprogesterone caproate, the 19-norprogesterone derivative nomegestrol acetate, and the 19-nortestosterone derivatives norethisterone acetate and norethisterone enanthate.
Progestogen esters

Estrogens were discovered in 1929,[4] and beginning in 1936, a variety of estradiol esters, such as estradiol benzoate and estradiol dipropionate, were introduced for clinical use.[4][5] Testosterone esters, such as testosterone propionate and testosterone phenylacetate, were also introduced around this time.[6] In contrast to estradiol and testosterone, progesterone proved more difficult to esterify.[7][8] In fact, esterification involves the replacement of a hydroxyl group with an alkoxy group,[9] and unlike estradiol and testosterone, progesterone does not possess any hydroxyl groups,[10] so it is actually not chemically possible to esterify progesterone itself.[8][11] The first progestogen esters were not introduced until the mid-1950s,[4][7][12] and were esters of 17α-hydroxyprogesterone (which, unlike progesterone, has a hydroxyl group available for esterification) rather than of progesterone; they included 17α-hydroxyprogesterone caproate (Delalutin, Proluton) and 17α-hydroxyprogesterone acetate (Prodrox).[2][12] The following quote of de Médicis Sajous et al. (1961) details the development of progestogen esters:[13]
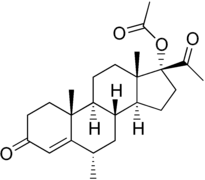
Over a period of several years, many tens of thousands of dollars were invested by Upjohn in an effort to find an easily absorbed, orally active progesterone ester. The effort met with but limited success. One promising ester, [17α-hydroxyprogesterone acetate], marketed as Prodox, was found. It was more active by mouth than other progesterone preparations then on the market, but it was not so active orally as desired. To obtain a progestational drug with the wanted properties, it appeared necessary to alter the progesterone molecule itself. Beginning about 1957, Upjohn steroid chemists accordingly prepared a series of progesterones modified in the various ways that had been found to multiply the power of cortisone and hydrocortisone. One of the modifications — worked out by a team under Dr. John C. Babcock — was the attachment of a carbon atom and three hydrogen atoms — a methyl group — to carbon 6 in the first ring of the progesterone steroid nucleus. A similar modification had been the key step in creating Medrol, Upjohn's high-potency, antiinflammatory cortisone-type steroid. The new progestational agent was [6α-methyl-17α-hydroxyprogesterone acetate] or [medroxyprogesterone acetate], which Upjohn has trademarked Provera. It has proved to be the most potent progestational drug yet uncovered — hundreds of times more active orally than progesterone and, weight for weight, some fifty times more active by subcutaneous injection. Provera was placed on the market in 1959.
Medroxyprogesterone acetate (Provera) entered clinical use and became widely marketed, largely superseding the 17α-hydroxyprogesterone esters.[4] A variety of analogues of medroxyprogesterone acetate, such as chlormadinone acetate, cyproterone acetate, and megestrol acetate, were subsequently developed and introduced as well.[2][4][14] Progestogen esters of other groups of progestins have also been introduced, including the 19-norprogesterone derivatives gestonorone caproate, segesterone acetate (nestorone), nomegestrol acetate, and norgestomet (11β-methyl-17α-acetoxy-19-norprogesterone) and the 19-nortestosterone derivatives etynodiol diacetate, norethisterone acetate, norethisterone enanthate, and quingestanol acetate.
Although esters of steroidal androgens and estrogens are generally inactive themselves and act as prodrugs, the same is not true for many progestogen esters. For instance, esters of 17α-hydroxyprogesterone derivatives, such as hydroxyprogesterone caproate, medroxyprogesterone acetate, and cyproterone acetate, are highly active themselves (in fact, they are far more active than their unesterified forms) and are not prodrugs, forming little or none of their parent compounds (in the cases of the examples given, hydroxyprogesterone, medroxyprogesterone, and cyproterone, respectively).[15][16] On the other hand, esters of 19-nortestosterone derivatives, such as etynodiol diacetate, norethisterone acetate, norethisterone enanthate, and quingestanol acetate, are all prodrugs.[17]
Progestogen ethers
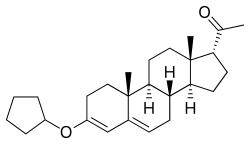

Although it cannot be esterified, progesterone possesses ketone groups at the C3 and C20 positions, and for this reason, it is possible to etherify it; that is, progesterone ethers are possible. Quingestrone (Enol-Luteovis) is a progesterone ether (specifically, the 3-cyclopentyl ether of progesterone) that has been marketed in Italy as an oral contraceptive.[18][19] Quingestrone is a variant of progesterone with improved pharmacokinetics, including higher potency, oral activity, greater lipophilicity, and a longer half-life.[20][21][22][23][24] Two other progestogens, pentagestrone (never marketed) and pentagestrone acetate (Gestovis, Gestovister), are the 3-cyclopentyl enol ethers of 17α-hydroxyprogesterone and 17α-hydroxyprogesterone acetate, respectively, while progesterone 3-acetyl enol ether (never marketed) is the 3-acetyl enol ether of progesterone.[3][18][25][26][27]
Although it was originally thought that progesterone ethers like quingestrone were prodrugs of progesterone, it was subsequently found that this is not the case and that quingestrone instead seems to be transformed directly into the corresponding alcohols rather than ketones.[28] These alcohols are progesterone metabolites like pregnanolones and pregnanediols, and as some of these metabolites, for instance 3β-dihydroprogesterone, have potent progestogenic activity, this may account for the clinical efficacy of progestogen ethers like quingestrone as progestogens.[28][29][27]
Progestogen oximes
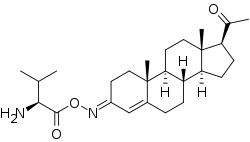
While not esters, C3 and C20 oxime conjugates of progesterone, such as progesterone carboxymethyloxime (progesterone 3-(O-carboxymethyl)oxime; P4-3-CMO), P1-185 (progesterone 3-O-(L-valine)-E-oxime), EIDD-1723 (progesterone (20E)-20-[O-[(phosphonooxy)methyl]oxime] sodium salt), EIDD-036 (progesterone 20-oxime), and VOLT-02 (chemical structure unreleased), have been developed as water-soluble progesterone and neurosteroid prodrugs, although none have completed clinical development or been marketed as of yet.[30][31][32][33][34][35]
Some 19-nortestosterone progestins, including the marketed progestins norgestimate and norelgestromin and the non-marketed progestin norethisterone acetate oxime, are C3 oximes, although they have potent progestogenic activity of their own and are not necessarily prodrugs of the corresponding ketones.[36]
See also
References
- Fraser, Ian S. (1998). Estrogens and Progestogens in Clinical Practice. Churchill Livingstone. p. 13. ISBN 978-0-443-04706-0.
- Lobo, Roger; Crosignani, P.G.; Paoletti, Rodolfo (31 October 2002). Women's Health and Menopause: New Strategies – Improved Quality of Life. Springer Science & Business Media. pp. 91–. ISBN 978-1-4020-7149-2.
- Korolkovas, Andrejus (16 August 1988). Essentials of Medicinal Chemistry. Wiley. p. 1021. ISBN 978-0-471-88356-2.
- Ravina, Enrique (11 January 2011). The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs. John Wiley & Sons. pp. 174–175, 194. ISBN 978-3-527-32669-3.
- Roche Review ... Hoffman-La Roche, and Roche-organon. 1944.
- Korenchevsky V, Dennison M, Eldridge M (1937). "The prolonged treatment of castrated and ovariectomized rats with testosterone propionate". Biochem. J. 31 (3): 475–85. doi:10.1042/bj0310475. PMC 1266958. PMID 16746360.
- Charles Eucharist de Medicis Sajous (1939). Analytic cyclopedia of practical medicine. Davis. p. 75.
- Boschann HW (July 1958). "Observations of the role of progestational agents in human gynecologic disorders and pregnancy complications". Ann N Y Acad Sci. 71 (5): 727–52. Bibcode:1958NYASA..71..727B. doi:10.1111/j.1749-6632.1958.tb46803.x. PMID 13583829.
In order to obtain the picture of a normal secretory phase, ten 20-mg. doses of progesterone daily are required (Ober and Weber, 1951). Corner (1947) supposes that this quantity is formed daily by the woman during the normal luteal phase. The results of experiments with 1 injection of 250 mg. and 2 injections each of 125 mg. of progesterone indicate that it is not possible to obtain an increase of effect and prolongation of action by raising the dose. Progesterone in oily solution is too rapidly excreted to develop an effect on the endometrium lasting longer than 48 hours (Bradbury et al., 1950; Zander, 1952). This is also reported by investigations on the blood level for the Hooker-Forbes test. Excessive quantities are excreted by the overflow effect (Schoeller and Gehrke, 1938). Crystalline pressings have not proved themselves in practice as implant tablets. In theory their absorption properties are good, but they are limited by defense reaction on the part of the organism. The quantity absorbed is not adequate for a therapeutic effect (approximately mg. daily from 100-mg. pressing). The enolic esters of progesterone are oxygen-sensitive and therefore unstable (Junkmann, 1954). It is not possible, therefore, to obtain depot preparations by the method of esterification as in the case of estrogens and androgens. Accordingly, Hohlweg, in 1953, wrote that no progesterone compounds with prolonged action were known.
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "esters". doi:10.1351/goldbook.E02219
- Nuclear Receptor Coregulators. Academic Press. 11 August 2004. pp. 69–. ISBN 978-0-08-052288-3.
- Bishop, P. M. F. (1958). "Endocrine Treatment of Gynaecological Disorders". In Gardiner-Hill, H. (ed.). Modern Trends in Endocrinology. Modern Trends. Vol. 1. London: Butterworth & Co. pp. 231–244.
- Sneader, Walter (23 June 2005). Drug Discovery: A History. John Wiley & Sons. pp. 204–. ISBN 978-0-471-89979-2.
- Engel, Leonard (1961). Medicine Makers of Kalamazoo. McGraw-Hill. p. 125.
- Shoupe, Donna (7 November 2007). The Handbook of Contraception: A Guide for Practical Management. Springer Science & Business Media. pp. 103–. ISBN 978-1-59745-150-5.
- Attardi BJ, Zeleznik A, Simhan H, Chiao JP, Mattison DR, Caritis SN (2007). "Comparison of progesterone and glucocorticoid receptor binding and stimulation of gene expression by progesterone, 17-alpha hydroxyprogesterone caproate, and related progestins". Am. J. Obstet. Gynecol. 197 (6): 599.e1–7. doi:10.1016/j.ajog.2007.05.024. PMC 2278032. PMID 18060946.
- Weber, Georg F. (22 July 2015). Molecular Therapies of Cancer. Springer. pp. 316–. ISBN 978-3-319-13278-5.
- Roberts, Stanley M.; Price, Barry J. (1985). Medicinal chemistry: the role of organic chemistry in drug research. Academic Press. ISBN 978-0-12-589730-3.
- Elks, J. (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 886–887, 943, 1058. ISBN 978-1-4757-2085-3.
- International Planned Parenthood Federation. Medical Committee. Oral Advisory Group (1965). Handbook on oral contraception. Little, Brown. p. 18.
- Burton, Eunice R.; Wachtel, Erica G. (1967). "A Clinical Trial and Cytological Assessment of Enol Luteovis in the Treatment of Threatened and Recurrent Abortion". BJOG: An International Journal of Obstetrics and Gynaecology. 74 (4): 533–536. doi:10.1111/j.1471-0528.1967.tb03986.x. PMID 5340429. S2CID 31602503.
- Charman, William N.; Porter, Christopher J.H. (1996). "Lipophilic prodrugs designed for intestinal lymphatic transport". Advanced Drug Delivery Reviews. 19 (2): 149–169. doi:10.1016/0169-409X(95)00105-G.
- Joseph Bolivar De Lee (1965). The ... Year Book of Obstetrics and Gynecology. Year Book Publishers. p. 150.
- Bentley, P. J. (1980). Endocrine Pharmacology: Physiological Basis and Therapeutic Applications. CUP Archive. pp. 274–. ISBN 978-0-521-22673-8.
- Current Medicine and Drugs. 1962.
Enol Luteovis (3 cyclo-pentyl enol ether of progesterone) is the only oral progestin producing pregnanediol as a metabolite. It is not very potent and probably carries very little risk of producing virilizing effects on a female foetus. Thus it is more closely related to progesterone than the other synthetic progestins.
- Wermuth, Camille Georges (2 May 2011). The Practice of Medicinal Chemistry. Academic Press. pp. 731–. ISBN 978-0-08-056877-5.
- Lutwak-Mann, Cecilia; Adams, C. E. (April 1957). "Carbonic anhydrase in the female reproductive tract. II. Endometrial carbonic anhydrase as indicator of luteoid potency: correlation with progestational proliferation". J. Endocrinol. 15 (1): 43–55. doi:10.1677/joe.0.0150043. PMID 13439082.
- Pincus G, Miyake T, Merrill AP, Longo P (November 1957). "The bioassay of progesterone". Endocrinology. 61 (5): 528–33. doi:10.1210/endo-61-5-528. PMID 13480263.
- Meli, A.; Wolff, A.; Lucker, W. E.; Steinetz, B. G. (1965). "The Biological Profile of Progesterone 3-Cyclopentyl Enol Ether as Compared with That of Progesterone". Experimental Biology and Medicine. 118 (3): 714–717. doi:10.3181/00379727-118-29947. PMID 14264537. S2CID 11891451.
- Junkermann H, Runnebaum B, Lisboa BP (July 1977). "New progesterone metabolites in human myometrium". Steroids. 30 (1): 1–14. doi:10.1016/0039-128X(77)90131-3. PMID 919010. S2CID 28420255.
In the Clauberg bioassay the 3β-hydroxy-4-pregnen-20-one shows about the same potency as progesterone (34). In regard to the biological activity of the 3α epimer no data are available.
- Basu, Krishnakali; Mitra, Ashim K. (1990). "Effects of 3-hydrazone modification on the metabolism and protein binding of progesterone". International Journal of Pharmaceutics. 65 (1–2): 109–114. doi:10.1016/0378-5173(90)90015-V.
- Wali B, Sayeed I, Guthrie DB, Natchus MG, Turan N, Liotta DC, Stein DG (October 2016). "Evaluating the neurotherapeutic potential of a water-soluble progesterone analog after traumatic brain injury in rats". Neuropharmacology. 109: 148–158. doi:10.1016/j.neuropharm.2016.05.017. PMID 27267687. S2CID 19906601.
- Guthrie, D. B., Lockwood, M. A., Natchus, M. G., Liotta, D. C., Stein, D. G., & Sayeed, I. (2017). "Progesterone phosphate analogs and uses related thereto" U.S. Patent 9,802,978.
- MacNevin CJ, Atif F, Sayeed I, Stein DG, Liotta DC (October 2009). "Development and screening of water-soluble analogues of progesterone and allopregnanolone in models of brain injury". J. Med. Chem. 52 (19): 6012–23. doi:10.1021/jm900712n. PMID 19791804.
- Guthrie DB, Stein DG, Liotta DC, Lockwood MA, Sayeed I, Atif F, Arrendale RF, Reddy GP, Evers TJ, Marengo JR, Howard RB, Culver DG, Natchus MG (May 2012). "Water-soluble progesterone analogues are effective, injectable treatments in animal models of traumatic brain injury". ACS Med Chem Lett. 3 (5): 362–6. doi:10.1021/ml200303r. PMC 4025794. PMID 24900479.
- Progesterone conjugate - Levolta Pharmaceuticals. springer.com
- Stanczyk FZ (November 2003). "All progestins are not created equal". Steroids. 68 (10–13): 879–90. doi:10.1016/j.steroids.2003.08.003. PMID 14667980. S2CID 44601264.
External links
 Media related to Progestogen esters at Wikimedia Commons
Media related to Progestogen esters at Wikimedia Commons