Équation d'état de van der Waals
En physique, et plus particulièrement en thermodynamique, l’équation d'état de van der Waals est une équation d'état des fluides réels proposée par le physicien Johannes Diderik van der Waals en 1873[1],[2]. Elle lui valut le prix Nobel de physique en 1910 « pour ses travaux sur l'équation d'état des gaz et des liquides[3]. »
La contribution fondamentale de van der Waals fut de modifier la loi des gaz parfaits en introduisant phénoménologiquement la taille finie des molécules ainsi que l'interaction attractive entre celles-ci. Le modèle des gaz parfaits, en effet, ne considère les particules que comme des sphères dures n'interagissant entre elles que par collision. Cette équation d'état peut également être établie à partir de la physique statistique[4]. Concernant la taille des molécules, on peut déduire de l'équation de van der Waals le rayon de van der Waals. Les interactions à distance entre molécules sont appelées forces de van der Waals.
L'équation d'état de van der Waals fut historiquement une avancée considérable par rapport à l'équation des gaz parfaits, puisque, en plus de décrire le comportement d'un gaz réel plus précisément que le modèle des gaz parfaits (notamment les variations de température lors d'une détente de Joule-Thomson), cette équation décrit qualitativement la transition de phase liquide-gaz et présente un point critique. Elle guida Kamerlingh Onnes dans ses travaux sur la liquéfaction de l'hélium (1908)[5] et fut à l'origine d'une véritable course au froid dans le but d'atteindre le zéro absolu[6].
Grâce à sa simplicité d'utilisation, cette équation fut modifiée et améliorée à de nombreuses reprises, elle est la première d'une famille d'équations appelées équations d'état cubiques, parmi lesquelles on peut citer les plus connues : celle de Redlich-Kwong (1949), modifiée par Soave (1972), et celle de Peng-Robinson (1976).
Équation de van der Waals
Forme extensive
L'équation d'état de van der Waals s'écrit sous forme extensive :

ou explicitement en pression :

avec :
- le terme de cohésion (constant) ;
- le covolume molaire (constant) ;
- la quantité de matière (nombre de moles) ;
- la pression ;
- la constante universelle des gaz parfaits ;
- la température absolue ;
- le volume.







Les expressions des paramètres de l'équation de van der Waals en fonction des pression, température et volume molaire critiques sont données plus bas dans le paragraphe Les paramètres a et b. Si l'on considère le nombre de molécules à la place du nombre de moles :

avec :
- ;
- le volume d'exclusion d'une molécule ;
- la constante de Boltzmann ;
- le nombre de molécules, ;
- le nombre d'Avogadro.






Rappelons que la constante universelle des gaz parfaits vaut .

Forme intensive
L'équation d'état peut aussi s'écrire sous forme intensive :

avec le volume molaire : .


Forme polynomiale
En multipliant par la forme intensive, l'équation de van der Waals s'écrit également sous la forme d'un polynôme de degré trois en , le facteur de compressibilité. Historiquement, elle est la première des équations d'état cubiques :



avec :
- le terme de cohésion normé ;
- le covolume molaire normé ;
- le facteur de compressibilité.



En comparaison, la loi des gaz parfaits revient à écrire .

Sous cette forme, l'équation d'état est facilement utilisable en employant la méthode de Cardan pour trouver les racines du polynôme. Lorsque l'équation d'état, à , et composition données, donne une seule racine réelle, il peut s'agir soit d'un gaz, soit d'un liquide. Si l'équation d'état donne trois racines réelles, la plus grande, , permet de calculer le volume molaire d'un gaz, , selon : ; la plus petite, , permet de calculer le volume molaire d'un liquide, , selon : ; la racine intermédiaire n'a pas de réalité physique (voir le paragraphe Isothermes d'Andrews, diagramme de Clapeyron).






Forme réduite
Selon la loi des états correspondants de van der Waals, le point critique joue un rôle important pour les gaz réels. L'équation de van der Waals peut enfin se réécrire sous forme réduite :

en introduisant les coordonnées réduites :
- la pression réduite ;
- la température réduite ;
- le volume réduit ;



avec :
- la pression critique ;
- la température critique ;
- le volume molaire critique.



Les expressions des pression, température et volume critiques en fonction des paramètres et de l'équation de van der Waals sont données plus bas dans le paragraphe Le point critique. Sous sa forme réduite, l'équation de van der Waals est indépendante de la nature du produit ; elle permet ainsi d'étudier l'équation d'état sans se rapporter à un produit spécifique, par exemple pour tracer les isothermes des figure 1 et figure 4.
Paramètres pour un corps pur
Pour un corps pur, les paramètres et sont calculés à partir des pression et température critiques (mesurables expérimentalement) selon :

avec :
- la pression critique du corps pur ;
- la température critique du corps pur ;
- la constante universelle des gaz parfaits.
Paramètres pour un mélange
Dans le cas d'un mélange de corps, les paramètres et sont calculés classiquement selon les règles de mélange suivantes, proposées par van der Waals en 1890[7] :

avec :
- la fraction molaire du corps ;
- le paramètre de l'équation de van der Waals pour le mélange ;
- le paramètre de l'équation de van der Waals pour le corps pur ;
- un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec et ;
- le paramètre de l'équation de van der Waals pour le mélange ;
- le paramètre de l'équation de van der Waals pour le corps pur.










Il existe d'autres règles de mélange, plus complexes et faisant intervenir d'autres paramètres. L'une des variantes les plus simples des règles de mélange classiques consiste à calculer le covolume selon :

avec un paramètre d'interaction binaire entre le corps et le corps , déterminé expérimentalement, avec et . Si tous les , cette règle revient à la règle classique.




Approche phénoménologique
En pratique, les gaz ne se comportent pas exactement comme le décrit la loi des gaz parfaits, car ils sont composés de molécules ayant un certain volume. Le volume accessible à une particule est alors inférieur au volume de l'enceinte. La réduction du volume accessible est proportionnelle au nombre total de particules (ou nombre de moles). Le volume idéal de l'équation d'état des gaz parfaits, correspondant au volume total de l'enceinte puisque les particules y sont supposées de volume nul, est ainsi remplacé par :

En outre, des interactions autres que les simples chocs élastiques du modèle des gaz parfaits existent entre les molécules. Les forces attractives entre les molécules font que la pression d'un gaz réel est inférieure à la pression d'un gaz idéal. L'équation de van der Waals s'obtient, contrairement à l'équation des gaz parfaits, à partir d'un modèle de gaz composé de sphères dures soumises à des interactions dipolaires attractives appelées forces de van der Waals, ce qui conduit à l'adaptation suivante :

En introduisant ces grandeurs dans l'équation des gaz parfaits :

on obtient l'équation de van der Waals :

On remarque qu'à quantité de matière constante lorsque le volume devient très grand on a :


on retrouve l'équation d'état du gaz parfait :
ce qui est cohérent avec le comportement des gaz réels.
Justification par la thermodynamique statistique
La fonction de partition (à ne pas confondre avec le facteur de compressibilité, également noté ) d'un gaz parfait constitué de particules identiques dans un volume est :


où est la longueur d'onde thermique de de Broglie :


avec :
- la constante de Planck ;
- la masse d'une particule ;
- la constante de Boltzmann ;
- la température ;
- la fonction de partition à une particule dans le volume .



Pour les gaz réels, les particules sont considérées comme indépendantes, la relation est conservée pour l'ensemble de particules, mais la fonction de partition d'une particule est modifiée par les interactions de cette particule avec les autres particules. L'interaction entre deux particules est modélisée par un potentiel de Sutherland :


avec :
- la distance entre les particules (entre les centres des sphères dures) ;
- la distance à laquelle les particules sont en contact (le double de leur rayon) ;
- l'intensité de la force de van der Waals entre les particules.



Premièrement, le volume occupé par l'ensemble des autres particules limite le volume accessible à une particule dans une enceinte de volume . Les particules sont considérées comme des sphères dures toutes identiques de diamètre ; le volume d'une particule est donc égal à . Lorsque deux particules sont en contact, leurs centres sont à une distance l'un de l'autre égale à la somme du rayon de la particule 1 et du rayon de la particule 2, soit un diamètre . Puisque les particules sont des sphères dures, les deux centres ne peuvent se situer à une distance inférieure à (interaction entre particules quand ).



On choisit arbitrairement une particule cible et une seconde particule impactant la première. Lors d'une collision dans l'espace à trois dimensions, il existe ainsi autour de la particule cible une sphère d'exclusion de rayon et de volume : dans laquelle le centre de la particule impactante ne peut pénétrer. Le choix de la particule cible et de la particule impactante étant arbitraire, il faut rapporter le volume d'exclusion aux deux particules ; le volume d'exclusion rapporté au nombre de particules en collision est de :


n'est donc pas le volume d'une particule : . Pour un ensemble de particules, le volume d'exclusion total est de . Le volume disponible pour une particule est ainsi corrigé en .




Deuxièmement, la prise en compte des interactions entre particules est effectuée en calculant la fonction de partition d'une particule sensible au champ moyen créé par l'ensemble des autres particules. La concentration de particules dans un volume est égale à , elle est considérée comme homogène dans tout le volume. On considère d'autre part une particule cible quelconque ; à une distance comprise entre et du centre de cette particule cible se trouvent particules. L'énergie d'interaction entre ces particules et la particule cible est de . Sur l'ensemble du volume , l'énergie d'interaction d'une particule avec le reste du gaz est égale à :






avec . L'intégration est effectuée à partir de car, comme vu plus haut, il ne peut pas se trouver d'autre particule à une distance plus petite du centre de la particule cible.


La fonction de partition à une particule fait alors intervenir un facteur de Boltzmann . Le facteur 2 vient du partage de l'énergie d'interaction entre les deux particules concernées.

On obtient ainsi la fonction de partition corrigée pour une particule :

puis la fonction de partition pour l'ensemble des particules :

La pression dans l'ensemble canonique se calcule à partir de :

La dérivation des seuls termes impliquant le volume donne alors l'équation d'état de van der Waals :



avec :
- la pression du gaz ;
- la température absolue ;
- le volume ;
- le covolume molaire ;
- le volume d'exclusion d'une particule ;
- le terme de cohésion ;
- ;
- la constante universelle des gaz parfaits, ;
- le nombre de molécules ;
- la quantité de matière ou nombre de moles de gaz, ;
- le nombre d'Avogadro ;
- la constante de Boltzmann.





Étude de l'équation d'état de van der Waals
Isothermes d'Andrews, diagramme de Clapeyron
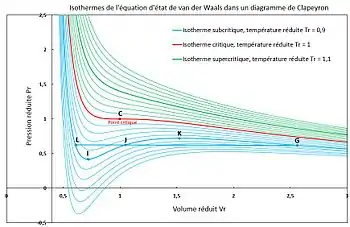
En rouge : l'isotherme critique. Au-dessus (courbes vertes) le fluide est supercritique. Au-dessous (courbes bleues) le fluide peut être liquide ou gazeux.
Pour tout corps pur réel, à température donnée, on peut tracer l'évolution de la pression en fonction du volume (à quantité de matière constante), ou du volume molaire (un diagramme portant le volume en abscisse et la pression en ordonnée est appelé diagramme de Clapeyron) : cette courbe est appelée courbe isotherme ou isotherme d'Andrews. Pour un gaz parfait, la loi de Boyle-Mariotte indique que la courbe isotherme est une branche d'hyperbole, puisque . La figure 1 montre sur le même graphique le tracé de plusieurs courbes isothermes obtenues pour un corps pur avec l'équation de van der Waals en coordonnées réduites :



Mathématiquement, les isothermes de l'équation d'état de van der Waals peuvent être tracées pour des volumes réduits allant de à . Cependant, du point de vue de la théorie physique, les volumes réduits ne peuvent être inférieurs à selon cette équation. La droite verticale d'équation constitue une asymptote pour toutes les isothermes. De ce fait, les isothermes ne sont étudiées que dans le domaine des volumes réduits supérieurs à .





L'étude de ces courbes montre l'existence d'une température critique (courbe rouge de la figure 1) telle que :
- pour une température l'équation de van der Waals n'associe qu'un seul volume à une pression donnée (courbes vertes de la figure 1) : l'équation d'état cubique ne produit qu'une seule racine réelle (un seul volume molaire) quelle que soit la valeur de la pression ;
- pour une température , le comportement est plus complexe (courbes bleu turquoise de la figure 1) : l'équation d'état cubique peut produire, selon la pression, une seule racine réelle (un seul volume molaire) ou trois racines réelles (trois volumes molaires).


Ces courbes sont interprétées comme suit :
- pour une température le fluide n'est stable que sous une seule phase : le fluide supercritique ;
- pour une température :
- le fluide est stable sous une seule phase, gaz ou liquide, lorsque l'équation d'état ne produit qu'une seule racine réelle :
- le fluide peut être présent simultanément sous deux phases en équilibre, gaz et liquide, lorsque l'équation d'état produit trois racines réelles.
Le point critique
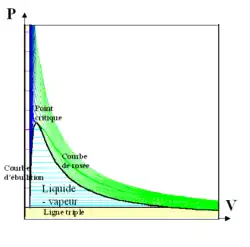
Ces isothermes expérimentales permettent entre autres d'observer la transition critique et les paliers de pression au changement de phase liquide-gaz.
La figure 2 montre l'allure des isothermes d'Andrews expérimentales. Dans ce diagramme, qui trace l'évolution de la pression en fonction du volume à température constante, une courbe isotherme à basse température est décroissante aux faibles volumes et fortes pressions, correspondant à un liquide, puis atteint un palier de pression constante, correspondant à un changement de phase liquide-vapeur, puis décroît pour de faibles pressions et de forts volumes, correspondant à un gaz. Les paliers de changement d'état sont donc horizontaux dans ce diagramme (paliers bleu turquoise de la figure 2). Plus la température augmente, plus la longueur du palier isobare correspondant diminue. Il existe une courbe, appelée isotherme critique, sur laquelle le palier est réduit à un point, appelé point critique (point C de la courbe rouge de la figure 1) : le liquide et le gaz ne s'y distinguent plus. Le point critique est donc un cas dégénéré de palier de changement d'état isobare. On en déduit que la tangente de la courbe isotherme critique en ce point doit être horizontale. De plus, puisque l'isotherme critique est décroissante monotone, elle traverse sa tangente en ce point : le point critique est un point d'inflexion. Au point critique la dérivée première (tangente horizontale) et la dérivée seconde (point d'inflexion) de la pression en fonction du volume, à température et quantité de matière constantes, sont par conséquent nulles[8],[9],[10] :

Remarque 1 - Les isothermes peuvent présenter plusieurs points d'inflexion[11]. Le point critique est le seul de toutes les isothermes où la tangente est horizontale dans le diagramme P-V.
Remarque 2 - La figure 2 est fausse, puisqu'au point critique la courbe critique y présente bien un point d'inflexion, mais que sa tangente n'y est pas horizontale ; la figure 1 est correcte de ce point de vue.
Avec l'équation d'état elle-même, on a un système de trois équations à trois inconnues (la pression, la température, le volume)[9],[8],[12] :



ou, en introduisant le volume molaire :



La solution, en introduisant les pression critique , température critique et volume molaire critique , est[9],[12] :

Note : cette solution est unique. Comme indiqué dans la remarque précédente, le point critique est bien l'unique point d'inflexion parmi tous les points d'inflexion de toutes les isothermes, supercritiques et subcritiques, dont la tangente a une pente nulle dans le diagramme P-V.
Les pression et température critiques étant mesurables expérimentalement, on peut établir la valeur des paramètres et [8],[12] :
Nous avons défini le facteur de compressibilité . Au point critique, l'équation de van der Waals donne pour ce paramètre la valeur[8],[12] :

Selon l'équation de van der Waals le facteur de compressibilité critique serait une constante universelle, mais cela ne correspond pas au comportement des gaz réels. En règle générale, le facteur de compressibilité critique vaut de 0,2 à 0,3[13],[12]. On a par exemple respectivement pour l'hydrogène, l'éthane, l'ammoniac et l'eau :


Les déviations observées montrent les limites de l'équation de van der Waals, qui ne peut représenter simultanément les trois propriétés critiques , et avec seulement deux paramètres, et . Toutefois, au regard de la loi des gaz parfaits selon laquelle quelles que soient les conditions de pression et de température, y compris au point critique, l'équation de van der Waals fut une avancée considérable dans la représentation des fluides et la compréhension des phénomènes physiques associés.

Certaines équations d'état dérivées de l'équation de van der Waals utilisent un troisième paramètre, , permettant de représenter simultanément les trois conditions du point critique. Cependant la mesure du volume molaire critique est difficile, étant donné les importantes fluctuations de la matière autour du point critique : le volume molaire critique est généralement mesuré avec une importante incertitude, ce paramètre n'est pas assez fiable pour être utilisé dans l'établissement d'une équation d'état.

Température de Boyle-Mariotte

En violet : isotherme de Boyle-Mariotte ; en tirets noirs : droite des gaz parfaits ; en rouge : isotherme critique.
L'équation d'état de van der Waals donne :


Pour de grands volumes molaires, , soit , on a par développement limité :



Ce qui aboutit à un développement de l'équation d'état de van der Waals sous forme d'une équation du viriel[14] :

Le deuxième coefficient du viriel vaut :

Il s'annule pour une température dite température de Boyle[14], ou température de Boyle-Mariotte, notée :


À cette température l'équation de van der Waals est au plus proche de l'équation d'état des gaz parfaits () : les forces d'attraction et de répulsion entre molécules de gaz s'y compensent exactement[15]. La courbe isotherme correspondante est tracée sur la figure 3. Cette figure montre également qu'à basse pression le comportement d'un gaz de van der Waals se rapproche de celui d'un gaz parfait quelle que soit la température.
Isothermes dans un diagramme d'Amagat, courbe de Boyle-Mariotte

En marron : courbe de Boyle-Mariotte ; en violet : isotherme de Boyle-Mariotte ; en rouge : isotherme critique.
Si l'on trace les isothermes d'un gaz parfait dans un diagramme d'Amagat, c'est-à-dire le produit de la pression et du volume en fonction de la pression pour différentes températures , on obtient des droites horizontales, puisque est une constante à température donnée : il s'agit de la loi de Boyle-Mariotte. Pour un gaz réel ces isothermes sont décroissantes aux basses pressions puis croissantes aux hautes pressions. La figure 4 représente les isothermes réduites de van der Waals dans un diagramme d'Amagat : cette équation d'état représente qualitativement le phénomène physique réel. Cette figure représente également l'isotherme de Boyle-Mariotte définie à la température de Boyle-Mariotte établie précédemment.
Le lieu géométrique des minimums des isothermes est appelé courbe de Boyle-Mariotte[16]. Cette courbe est donnée par la relation :

qui, puisque les coordonnées critiques sont des constantes, peut être écrite en coordonnées réduites :

que l'on peut développer et réarranger pour obtenir :

En utilisant la forme explicite réduite de l'équation de van der Waals :
on obtient :

en remplaçant et en réarrangeant :


La relation induit que :

On a donc :

que l'on multiplie par pour obtenir l'équation de la courbe de Boyle-Mariotte dans le diagramme d'Amagat :


Cette courbe est appelée ainsi car en chacun de ses points , comme pour un gaz parfait qui suit la loi de Boyle-Mariotte, à la différence près que pour le gaz parfait cette relation est vraie pour toute pression à toute température, donc en tout point du diagramme d'Amagat et non sur une unique courbe bien définie. Chacun des points de la courbe de Boyle-Mariotte est appelé point de Boyle-Mariotte de l'isotherme correspondante.
La courbe de Boyle-Mariotte intercepte l'isotherme de Boyle-Mariotte au point de coordonnées :
- ;
- , avec ;
- .




Au-delà de la température de Boyle-Mariotte les isothermes n'ont pas de point de Boyle-Mariotte et sont strictement croissantes dans le diagramme d'Amagat.
La courbe de Boyle-Mariotte intercepte l'axe des abscisses pour , soit . La forme réduite de l'équation d'état de van der Waals donne :



on retrouve la température de Boyle-Mariotte :

Le maximum de la courbe de Boyle-Mariotte a pour coordonnées :
- , soit ;
- , soit ;
- , soit .






On pose ; la courbe de Boyle-Mariotte a pour équation :


L'extremum se trouve en :


La courbe de Boyle-Mariotte donne :

et par conséquent :
L'équation de van der Waals donne :
Détente de Joule-Gay-Lussac
La détente de Joule-Gay-Lussac est une détente isoénergétique. Le coefficient de Joule-Gay-Lussac est défini par :

avec :


Il permet de quantifier le changement de température subi par le gaz dans cette détente. Pour un gaz parfait, selon la loi de Joule et Gay-Lussac, : un gaz parfait ne change pas de température dans une détente de Joule-Gay-Lussac.

Pour un gaz de van der Waals, on a :


On a donc : dans une détente de Joule-Gay-Lussac, un gaz de van der Waals ne peut que refroidir (lorsque son volume augmente à énergie constante sa température diminue). C'est le cas de la majorité des gaz, à l'exception notable de l'hélium, de l'hydrogène et de certains gaz rares qui se réchauffent sous certaines conditions de température initiale dans une détente de ce type[17].

Détente de Joule-Thomson
La détente de Joule-Thomson est une détente isenthalpique. Le coefficient de Joule-Thomson est défini par[18] :

avec :


Il permet de quantifier le changement de température subi par le gaz dans cette détente. Pour un gaz parfait, selon la loi de Joule-Thomson, : un gaz parfait ne change pas de température dans une détente de Joule-Thomson.

Pour un gaz de van der Waals, on a :


avec la capacité thermique isobare molaire. Il existe donc des températures pour lesquelles :




correspondant aux pressions :

On peut écrire, en coordonnées réduites :


et, puisque , on a la relation :


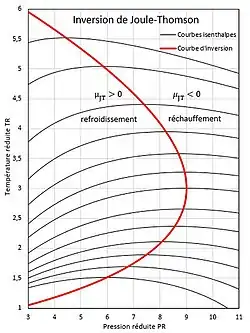
En rouge, la courbe d'inversion de Joule-Thomson. À gauche de la courbe d'inversion la température diminue lors d'une détente, à droite elle augmente[18].
Si l'on trace dans un diagramme les courbes isenthalpes d'un gaz de van der Waals (voir figure 5), certaines courbes présentent un maximum. Le lieu géométrique des maximums est la courbe définie ci-dessus, elle délimite deux domaines[18] :

- l'un dans lequel aux basses pressions : lors d'une détente de Joule-Thomson le gaz se refroidit (lors d'une diminution de pression à enthalpie constante la température diminue) ;
- l'autre dans lequel aux hautes pressions : lors d'une détente de Joule-Thomson le gaz se réchauffe (lors d'une diminution de pression à enthalpie constante la température augmente).


Un gaz ne peut donc être refroidi puis liquéfié[18],[19] :
- par détente que si ;
- par compression que si .
La pression ne pouvant être que positive, la courbe d'inversion admet deux limites à [18] :

- , soit ;
- , soit .




Un gaz ne peut donc être refroidi par détente de Joule-Thomson que si . Au-delà de un gaz se réchauffe dans une détente de Joule-Thomson quelle que soit la pression : les courbes isenthalpes sont strictement décroissantes, .


Le maximum de la courbe d'inversion a pour coordonnées :
- , soit ;
- , soit .




Pour tracer les courbes isenthalpes d'un gaz de van der Waals, il faut disposer d'un modèle d'enthalpie qui à priori dépend de la nature de ce gaz. Si l'on considère les relations :


on a :

alors est équivalent à . On a par définition :


avec :
- l'enthalpie du gaz réel à et ;
- l'enthalpie du gaz parfait correspondant à et ;
- l'enthalpie résiduelle à et .



Or, selon la loi de Joule-Thomson, l'enthalpie d'un gaz parfait ne dépend que de la température (aussi un gaz parfait ne change-t-il pas de température lors du changement de pression isenthalpe et dans le diagramme T-P les courbes isenthalpes d'un gaz parfait sont-elles des droites horizontales), d'où :

Par conséquent, les maximums des courbes isenthalpes sont identiques à ceux de l'enthalpie résiduelle. On peut donc étudier l'inversion de Joule-Thomson sur la seule composante résiduelle de l'enthalpie dont l'expression issue de l'équation d'état de van der Waals est :


La figure 5 est tracée en supposant la composante de l'enthalpie du gaz parfait nulle à toute température (). Les courbes isenthalpes sont données à enthalpie résiduelle réduite constante. Cette figure est donc totalement indépendante de la nature du gaz étudié.


Limites et défauts de l'équation

L'équation de van der Waals fut historiquement une avancée considérable par rapport à l'équation des gaz parfaits, puisqu'elle représente qualitativement le comportement des fluides réels aussi bien par la présence d'un point critique que par la représentation des phases liquide et gaz en équilibre et hors équilibre. Elle présente cependant certains défauts.
Représentation de la zone critique et de la phase liquide
Comme toutes les équations d'état cubiques, l'équation d'état de van der Waals ne peut prétendre représenter correctement à la fois la zone du point critique (voir paragraphe Le point critique) et le comportement des phases liquides. La comparaison de la figure 3, qui représente les isothermes de l'équation d'état de van der Waals dans un diagramme d'Amagat, et de la figure 6, qui représente les isothermes dans le même diagramme pour quelques corps réels, montre un large décalage, par exemple, pour l'isotherme critique : la courbe critique calculée a un minimum à environ , les valeurs réelles peuvent descendre à environ .


Il a été démontré que ceci était inhérent à la forme même de ces équations, aussi sont-elles destinées à représenter les phases gaz avant toute chose. Les volumes molaires liquides produits par des équations d'état cubiques peuvent présenter d'importants écarts (en excès le plus souvent) par rapport à la réalité, en particulier à basse pression.
Représentation des équilibres liquide-vapeur
Expérimentalement (voir figure 2), une courbe isotherme d'un corps pur aux fortes pressions et faibles volumes molaires décroît jusqu'à une pression appelée pression de vapeur saturante, notée : cette branche représente la phase liquide. L'isotherme passe ensuite par un palier à pression constante représentant le changement de phase liquide-gaz. L'isotherme redécroît ensuite aux faibles pressions et forts volumes molaires, représentant la phase gaz.

L'équation de van der Waals ne montre pas ce palier et suggère pour un corps pur la coexistence des deux phases liquide et gaz sur une certaine plage de pression pour une même température : selon les isothermes bleu turquoise de la figure 1, la coexistence des deux phases serait possible entre les pressions des points I et K.
L'équation de van der Waals est donc incorrecte sur la zone de coexistence des deux phases liquide et gaz et ne permet pas le calcul direct des pressions de vapeur saturante. La règle du palier de Maxwell permet de corriger ce défaut (voir paragraphe Pression de vapeur saturante).
Zone de métastabilité, zone d'instabilité et pressions négatives
Il est possible, à température donnée, dans certaines conditions très particulières, de maintenir de l'eau à l'état liquide sous une pression inférieure à sa pression de vapeur saturante (on parle de surchauffe ou retard à la vaporisation), de même qu'il est possible de maintenir de l'eau en phase gaz (vapeur d'eau) à une pression supérieure à sa pression de vapeur saturante (retard à la condensation). Ces états sont dits métastables et la moindre perturbation provoque l'évaporation violente de l'eau dans le premier cas et la liquéfaction brutale de la vapeur dans le second. Ces états sont similaires à celui de la surfusion dans lequel de l'eau est maintenue liquide en dessous de sa température de fusion et se solidifie à la moindre perturbation.
On peut également observer expérimentalement les pressions négatives obtenues pour la branche liquide des isothermes (isothermes bleu turquoise basses de la figure 1). Un tube de liquide placé dans une centrifugeuse subit une force qui peut être interprétée comme résultant d'une pression négative (ou tension)[20]. De l'eau refroidie dans une volume constant peut rester liquide à une température de −15 °C, produisant une pression de −1 200 bar[21]. On observe de l'eau liquide au sommet d'arbres de plus de 90 m de haut tel le Séquoia géant, alors que la capillarité ne peut faire monter l'eau à pression atmosphérique qu'à un maximum de 10 m : l'évaporation de l'eau dans les feuilles crée une pression de l'ordre de −4,8 atm dans des arbres de 60 m[22],[23].
Ces états métastables sont dus aux forces d'attraction entre molécules que l'équation de van der Waals introduit via le paramètre : en l'absence d'impuretés, de sites de nucléation, une phase peut se maintenir au-delà des conditions habituelles de changement de phase. La surface comprise entre la courbe de saturation et la courbe spinodale représentées sur la figure 7 correspond au domaine de métastabilité des isothermes de van der Waals. L'équation de van der Waals représente donc plus ou moins ces états métastables respectivement par les branches LI et KG des isothermes bleu turquoise de la figure 1, ce qui est à porter à son crédit. Mais elle présente également sur ces mêmes isothermes une branche IJK qui s'appliquerait à une phase dont le volume molaire augmenterait avec une augmentation de pression : cet état serait thermodynamiquement instable, car de compressibilité négative, et n'a jamais été observé, il n'a pas de sens physique[20]. La courbe spinodale représentée sur la figure 7 délimite le domaine d'instabilité des isothermes de van der Waals.

De façon générale, toute la partie des isothermes bleu turquoise comprise entre les points L et G (courbe LIJKG de la figure 1) n'est pas exploitable car peu, voire pas du tout, réaliste.
Absence de la phase solide
Si l'équation d'état présente bien un point critique, le fluide supercritique et la transition liquide-gaz, elle ne présente pas de point triple et ne dit en conséquence rien des transitions liquide-solide et gaz-solide. L'équation d'état ne représente pas la phase solide. Aux basses températures (en dessous de la température triple) elle n'est donc utilisable que pour représenter un gaz.
Courbes isochores
Si l'on trace la pression en fonction de la température à volume constant, les courbes isochores obtenues avec l'équation d'état de van der Waals sont des droites, ce qui n'est pas réaliste :

Ceci implique aussi que la capacité thermique isochore d'un fluide de van der Waals ne dépend pas du volume, ce qui n'est également pas réaliste (voir paragraphe Capacité thermique isochore). D'autres équations d'état dérivées de l'équation d'état de van der Waals, comme l'équation d'état de Redlich-Kwong, celle de Soave-Redlich-Kwong et celle de Peng-Robinson, pour ne citer que les plus connues et les plus utilisées, tentent de corriger ce défaut en faisant dépendre le paramètre de la température.
Grandeurs calculables à partir de l'équation d'état de van der Waals
Diverses grandeurs peuvent être calculées à partir de l'équation d'état de van der Waals, notamment la pression de vapeur saturante d'un corps pur, le coefficient de fugacité, les grandeurs résiduelles et les coefficients thermoélastiques.
Pression de vapeur saturante
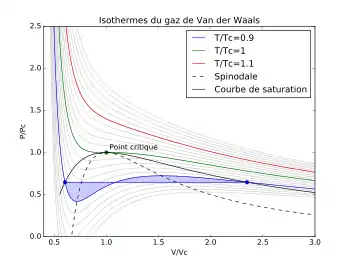
Ce qui suit n'est applicable qu'à un corps pur. Le calcul des pressions de bulle et de rosée d'un mélange est plus complexe et nécessite le calcul des coefficients de fugacité de chacun des composants du mélange.
Pour une température donnée inférieure à la température critique , pour des pressions inférieures à la pression critique , l'équation de van der Waals peut produire trois valeurs du volume molaire : le polynôme de degré trois en peut en effet avoir trois racines réelles. La plus grande de ces racines permet le calcul d'un volume molaire attribuable à un gaz et noté . La plus petite de ces racines permet de calculer un volume molaire attribuable à un liquide et noté . La racine intermédiaire n'a pas de sens physique, il s'agirait d'une phase dont le volume molaire augmenterait avec une augmentation de pression, ce qui n'existe pas.

Cependant, comme indiqué plus haut, l'équation de van der Waals suggère la coexistence des deux phases liquide et gaz sur une certaine plage de pression pour une même température pour un corps pur. Or l'expérience montre que pour un corps pur cette coexistence, ou équilibre, à température donnée, n'a lieu qu'à une seule pression : la pression de vapeur saturante notée .

La règle du palier de Maxwell indique que le gaz et le liquide sont à l'équilibre si :

Ce qui revient à dire que les deux surfaces violettes représentées sur la figure 7 sont égales.
L'équation d'état de van der Waals donne :

On a donc, en appliquant la règle du palier de Maxwell à l'équation d'état de van der Waals :

La température étant donnée, on a un système de trois équations à trois inconnues (les volumes molaires et et la pression ) :



Le calcul de la pression de vapeur saturante d'un corps pur à température donnée peut s'effectuer de façon itérative comme suit :
- fixer la pression ,
- calculer la ou les racines de la forme polynomiale cubique de l'équation d'état de van der Waals à et par la méthode de Cardan,
- si l'équation de van der Waals ne produit qu'une seule racine réelle, recommencer en 1,
- si l'équation de van der Waals produit trois racines réelles, calculer à partir de la plus grande et à partir de la plus petite,
- calculer ,
- si recommencer en 1,
- sinon .





Une fois la pression de vapeur saturante connue, on peut écarter les phases métastables et instables lorsque l'équation d'état cubique produit trois racines réelles à la même température mais à d'autres pressions que la pression de vapeur saturante. En dessous de seule la phase gaz existe de façon stable : si l'équation de van der Waals produit trois racines réelles seule celle attribuable à un gaz, la plus grande, doit être retenue. Au-dessus de seule la phase liquide est stable : si l'équation de van der Waals produit trois racines réelles seule celle attribuable à un liquide, la plus petite, doit être retenue.
Exemple - Pression de vapeur saturante de l'azote[24].
- Pour l'azote on dispose des données suivantes :
- température critique = 126,1 K ;
- pression critique = 35 bar.
- Pour une température de 100 K on calcule :
- = 0,133 Pa m6 mol−2 ;
- = 3,744 × 10−5 m3 mol−1 ;
- = 0,575 09 × 10−4 m3 mol−1 ;
- = 0,136 79 × 10−3 m3 mol−1 ;
- = 1,28 MPa.
- La littérature donne une pression de vapeur saturante expérimentale de l'azote à 100 K égale à 0,76 MPa. Ceci illustre les défauts de l'équation de van der Waals.
Coefficients pour un corps pur
Pour un corps pur (liquide ou gazeux) le coefficient de fugacité calculé avec l'équation d'état de van der Waals vaut :

ou, sous forme adimensionnelle :

avec :
- le terme de cohésion de l'équation de van der Waals ;
- le terme de cohésion normé ;
- le covolume molaire de l'équation de van der Waals ;
- le covolume molaire normé ;
- le volume molaire ;
- le facteur de compressibilité ;
- le coefficient de fugacité du corps pur.

On a donc, en fonction des grandeurs résiduelles molaires :

À saturation, les coefficients de fugacité du corps pur en phases liquide et vapeur sont égaux. On note :
- le coefficient de fugacité du corps pur en phase gaz ;
- le coefficient de fugacité du corps pur en phase liquide ;
- le volume molaire du corps pur en phase gaz ;
- le volume molaire du corps pur en phase liquide.


On a donc, à saturation :


En réarrangeant, on retrouve l'expression issue de la règle du palier de Maxwell appliquée à l'équation d'état de van der Waals pour le calcul de la pression de vapeur saturante :

Coefficients pour un corps dans un mélange
Dans ce qui suit un mélange (liquide ou gazeux) de corps est considéré. Le calcul du coefficient de fugacité d'un corps dans un mélange dépend des règles de mélange employées pour le calcul des paramètres et . Les expressions données ci-dessous ne sont valables qu'avec les règles de mélange classiques.
Le coefficient de fugacité de tout corps du mélange est calculé selon :

ou, sous forme adimensionnelle :

avec :
- le terme de cohésion normé pour le mélange ;
- le terme de cohésion normé pour le corps dans le mélange ;
- le covolume molaire normé pour le mélange ;
- le covolume molaire normé pour le corps dans le mélange ;
- le volume molaire du mélange ;
- le facteur de compressibilité du mélange ;
- ;
- le coefficient de fugacité du corps en mélange.






On a, par définition des règles de mélange classiques :



On peut écrire, en pondérant par les fractions molaires et en sommant sur l'ensemble des constituants du mélange :

On a donc, en fonction des grandeurs résiduelles molaires :

Grandeurs résiduelles
Les grandeurs résiduelles expriment l'écart entre les propriétés extensives d'un mélange réel et celles d'un mélange de gaz parfaits . Pour calculer une propriété d'un mélange réel (liquide ou gaz), il suffit donc de calculer la propriété correspondante d'un mélange de gaz parfaits et de lui ajouter la grandeur résiduelle appropriée, toutes ces propriétés étant calculées aux mêmes pression, température et composition que le mélange réel :




Les expressions données ci-dessous ne sont valables qu'avec les règles de mélange classiques. Pour un corps pur comme pour un mélange (liquide ou gazeux), les grandeurs résiduelles calculées avec l'équation d'état de van der Waals valent (les expressions sont données à chaque fois sous forme dimensionnelle et sous forme adimensionnelle) :
- énergie interne résiduelle molaire :

- volume résiduel molaire :

- enthalpie résiduelle molaire :

- entropie résiduelle molaire :

- énergie libre résiduelle molaire :

- enthalpie libre résiduelle molaire :

avec :
- le terme de cohésion de l'équation de van der Waals ;
- le terme de cohésion normé ;
- le covolume molaire de l'équation de van der Waals ;
- le covolume molaire normé ;
- le volume molaire ;
- le facteur de compressibilité.
Rappelons que par définition :
- pour un corps pur : ;
- pour un mélange : .


On remarquera également que pour un gaz parfait (corps pur ou mélange) , et : toutes les grandeurs résiduelles sont nulles comme il se doit.


Coefficients thermoélastiques
À partir de la forme extensive de l'équation d'état de van der Waals, on peut écrire :


d'où l'on tire l'expression du coefficient de dilatation isobare :

Toujours à partir de la forme extensive en pression de l'équation d'état, on a :

d'où l'on tire l'expression du coefficient de compression isochore :

Étant donné la relation :

on tire l'expression du coefficient de compressibilité isotherme :

En remplaçant dans ces expressions et en réarrangeant, on a :

Pour un gaz parfait et , on retrouve : , et .





Capacité thermique isochore
L'énergie interne molaire d'un fluide réel quelconque (gaz ou liquide, pur ou mélange) peut être décomposée en deux termes :


avec :
- l'énergie interne molaire du gaz parfait correspondant sous la même pression, à la même température et de même composition que le corps réel ;
- l'énergie interne résiduelle molaire, correspondant à l'écart entre le gaz parfait et le corps réel et due aux interactions entre les molécules du corps réel.


La capacité thermique isochore molaire vaut :

On définit de même :
- pour le gaz parfait ;
- pour la grandeur résiduelle.


Ce qui induit que :

Puisque, selon l'équation de van der Waals :

on a, le paramètre ne dépendant pas de la température dans l'équation d'état de van der Waals :

Ainsi, pour un fluide de van der Waals :

D'autre part, l'équation de van der Waals donne :

Pour un fluide de van der Waals, qu'il soit gaz ou liquide, la capacité thermique isochore du fluide réel est égale à celle du gaz parfait correspondant, et cette capacité ne varie pas avec le volume. Ceci n'est pas réaliste. D'autres équations d'état dérivées de l'équation d'état de van der Waals, comme l'équation d'état de Redlich-Kwong, celle de Soave-Redlich-Kwong et celle de Peng-Robinson, pour ne citer que les plus connues et les plus utilisées, tentent de corriger ce défaut en faisant dépendre le paramètre de la température.
Capacité thermique isobare
L'enthalpie molaire d'un fluide réel quelconque (gaz ou liquide, pur ou mélange) peut être décomposée en deux termes :


avec :
- l'enthalpie molaire du gaz parfait correspondant sous la même pression, à la même température et de même composition que le corps réel ;
- l'enthalpie résiduelle molaire, correspondant à l'écart entre le gaz parfait et le corps réel et due aux interactions entre les molécules du corps réel.


La capacité thermique isobare molaire vaut :

On définit de même :
- pour le gaz parfait ;
- pour la grandeur résiduelle.


Ce qui induit que :

Puisque, selon l'équation de van der Waals :
on a :

Ainsi, pour un fluide de van der Waals :

Pour un gaz parfait et , on retrouve : .

D'autre part, l'équation de van der Waals donne :
- en fonction de et :

- en fonction de et :

L'équation de van der Waals donne :




Si l'on remplace :


Si l'on remplace :

Pour un gaz parfait et , on retrouve : .

Selon l'équation d'état de van der Waals, la capacité thermique isobare est égale à la capacité isobare du gaz parfait correspondant corrigée par un terme résiduel additif dû aux interactions entre molécules. Ce terme résiduel induit une dépendance de la capacité thermique isobare à la pression, ce qui est plus réaliste que le modèle des gaz parfaits selon lequel, en vertu de la loi de Joule-Thomson, l'enthalpie du gaz parfait, et donc sa capacité isobare, ne dépend que de la température. Le terme résiduel induit également un écart entre la capacité thermique isobare d'un gaz et celle d'un liquide. Par exemple, un liquide et sa vapeur à l'équilibre (et par conséquent sous la même pression et à la même température) ont des volumes molaires différents, aussi les termes résiduels liés aux deux phases sont-ils différents : on obtient donc deux capacités thermiques isobares molaires différentes, bien que le terme lié au gaz parfait soit le même pour les deux phases. La représentation de la capacité thermique isobare par l'équation de van der Waals est donc plus réaliste que celle de la capacité thermique isochore.
Relation de Mayer
La relation de Mayer s'écrit :


avec :
Pour un gaz parfait et , on retrouve : .

Valeur des paramètres a et b pour divers corps
Les tableaux suivants[25] donnent la liste des paramètres et de van der Waals pour un certain nombre de gaz communs et de liquides volatils.
Unités :
- 1 J m3 mol−2 = 1 m6 Pa mol−2 = 10 l2 bar mol−2
- 1 m3 mol−1 = 1 000 l mol−1 = 1 000 dm3 mol−1
| Corps | (l2 bar mol−2) | (l mol−1) |
|---|---|---|
| Acétate d'éthyle | 20,72 | 0,1412 |
| Acétone | 14,09 | 0,0994 |
| Acétonitrile | 17,81 | 0,1168 |
| Acétylène | 4,448 | 0,05136 |
| Acide acétique | 17,82 | 0,1068 |
| Ammoniac | 4,225 | 0,03707 |
| Anhydride acétique | 20,16 | 0,1263 |
| Argon | 1,363 | 0,03219 |
| Azote | 1,408 | 0,03913 |
| Benzène | 18,24 | 0,1154 |
| Bromobenzène | 28,94 | 0,1539 |
| Bromure d'hydrogène | 4,510 | 0,04431 |
| Butane | 14,66 | 0,1226 |
| Chlore | 6,579 | 0,05622 |
| Chlorobenzène | 25,77 | 0,1453 |
| Chloroéthane | 11,05 | 0,08651 |
| Chlorofluorocarbure | 10,78 | 0,0998 |
| Chlorométhane | 7,570 | 0,06483 |
| Chlorure d'étain(IV) | 27,27 | 0,1642 |
| Chlorure d'hydrogène | 3,716 | 0,04081 |
| Cyanogène | 7,769 | 0,06901 |
| Cyclohexane | 23,11 | 0,1424 |
| Dioxyde d'azote | 5,354 | 0,04424 |
| Dioxyde de carbone | 3,640 | 0,04267 |
| Dioxyde de soufre | 6,803 | 0,05636 |
| Eau | 5,536 | 0,03049 |
| Éthane | 5,562 | 0,0638 |
| Éthanethiol | 11,39 | 0,08098 |
| Éthanol | 12,18 | 0,08407 |
| Éther diéthylique | 17,61 | 0,1344 |
| Éthylamine | 10,74 | 0,08409 |
| Fluorobenzène | 20,19 | 0,1286 |
| Fluorométhane | 4,692 | 0,05264 |
| Hélium | 0,03457 | 0,0237 |
| Hexane | 24,71 | 0,1735 |
| Hydrogène | 0,2476 | 0,02661 |
| Iodobenzène | 33,52 | 0,1656 |
| Krypton | 2,349 | 0,03978 |
| Mercure | 8,200 | 0,01696 |
| Méthane | 2,283 | 0,04278 |
| Méthanol | 9,649 | 0,06702 |
| Méthoxyméthane | 8,180 | 0,07246 |
| Monoxyde d'azote | 1,358 | 0,02789 |
| Monoxyde de carbone | 1,505 | 0,03985 |
| Néon | 0,2135 | 0,01709 |
| Oxygène | 1,378 | 0,03183 |
| Pentane | 19,26 | 0,146 |
| Phosphine | 4,692 | 0,05156 |
| Propane | 8,779 | 0,08445 |
| Protoxyde d'azote | 3,832 | 0,04415 |
| Séléniure d'hydrogène | 5,338 | 0,04637 |
| Silane | 4,377 | 0,05786 |
| Sulfure de carbone | 11,77 | 0,07685 |
| Sulfure de diéthyle | 19,00 | 0,1214 |
| Sulfure de diméthyle | 13,04 | 0,09213 |
| Sulfure d'hydrogène | 4,490 | 0,04287 |
| Tétrachlorométhane | 19,7483 | 0,1281 |
| Tétrachlorure de germanium | 22,90 | 0,1485 |
| Tétrafluorure de silicium | 4,251 | 0,05571 |
| Toluène | 24,38 | 0,1463 |
| Xénon | 4,250 | 0,05105 |
| Corps | (kPa dm6 mol−2) | (dm3 mol−1) | |
|---|---|---|---|
| Air (80 % N2, 20 % O2) | 135,8 | 0,0364 | |
| Ammoniac (NH3) | 422,4 | 0,0371 | |
| Argon (Ar) | 136,3 | 0,0322 | |
| Diazote (N2) | 140,8 | 0,0391 | |
| Dichlore (Cl2) | 657,4 | 0,0562 | |
| Dihydrogène (H2) | 24,7 | 0,0266 | |
| Dioxyde de carbone (CO2) | 363,7 | 0,0427 | |
| Dioxygène (O2) | 137,8 | 0,0318 | |
| Eau (H2O) | 557,29 | 0,031 | |
| Hélium (He) | 3,45 | 0,0237 | |
| Méthane (CH4) | 225 | 0,0428 | |
| Néon (Ne) | 21,3 | 0,0171 | |
| Données expérimentales sujettes à d'importantes variations. | |||
| Gaz | en (bar dm6 mol−2) | en (dm3 mol−1) | |
|---|---|---|---|
| Benzène (C6H6) | 52,74 | 0,3043 | |
| Décane (C10H22) | 37,88 | 0,2374 | |
| Octane (C8H18) | 18,82 | 0,1193 | |
| Données valables pour la phase gazeuse uniquement. | |||
Notes et références
- Cet article est partiellement ou en totalité issu de l'article intitulé « Liste des constantes de van der Waals » (voir la liste des auteurs).
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Van der Waals constants (data_page) » (voir la liste des auteurs).
Notes
- Extrait de la thèse de Johannes Diderik van der Waals : Over de Continuiteit van den gas- en vloeistoftoestand (De la continuité des états liquides et gazeux), Sijthoff, Leiden (Leyde), 1873.
- Johannes Diderik van der Waals (trad. de l'allemand), La Continuité des états gazeux et liquide, Georges Carré, , 279 p. (lire en ligne).
- (en) « For his work on the equation of state for gases and liquids. » in Personnel de rédaction, « The Nobel Prize in Physics 1910 », Fondation Nobel, 2010. Consulté le 17 avril 2016.
- Bernard Diu, Claudine Guthmann, Danielle Lederer et Bernard Roulet, Physique statistique, Paris, Éditions Hermann, , 1001 p. (ISBN 2-7056-6065-8).
- La première liquéfaction de l’hélium par Heike Kamerlingh Onnes, Benjamin Bradu, ingénieur au CERN, BibNum.
- Le zéro absolu et la course au froid, Université du Québec à Chicoutimi (UQAC).
- (de) J. D. van der Waals, « Molekulartheorie eines Körpers, der aus zwei verschiedenen Stoffen besteht » [« Théorie moléculaire pour un corps composé de deux espèces chimiques différentes »], Zeitschrift für Physikalische Chemie, vol. 5U, no 1, , p. 133–173 (ISSN 2196-7156 et 0942-9352, DOI 10.1515/zpch-1890-0514, lire en ligne, consulté le ).
- Vidal 1997, p. 34;114-115.
- Zemansky et al. 1997, p. 232-233.
- Corriou 1985, p. 2.
- L'équation n'a de solution en que si : une si , deux si . En substituant , on a .
- Roland Solimando, Louis Schuffenecker et Jean-Noël Jaubert, Propriétés thermodynamiques du corps pur, Éditions techniques de l'ingénieur (no AF 4 050), (lire en ligne), p. 9.
- Bernard Le Neindre, Constantes physiques des fluides purs : méthodes d’estimation, vol. K 692, éd. Techniques de l'Ingénieur, (lire en ligne), p. 16.
- (en) David W. Ball, Physical Chemistry, Cengage Learning, , 880 p. (ISBN 978-1-285-96977-0, lire en ligne), p. 13-15.
- Hervé Guérin, « L'équation de van der Waals et le viriel », Bulletin de l'Union des physiciens, Union des Professeurs de Physique et de Chimie (UdPPC), vol. 87, no SPÉCIAL, , p. 41-59 (lire en ligne, consulté le ).
- Thermodynamique et énergétique, Volume 1, Lucien Borel, Daniel Favrat, Presses Polytechniques et Universitaires Romandes, 2005, (ISBN 2-88074-545-4), p. 264.
- J.-Ph. Qadri (Académie de Bordeaux), « T4 – Appendice 1 - Détentes de Joule - Bilans énergétique et entropique » [PDF], sur webetab.ac-bordeaux.fr (consulté le ), p. 2-3.
- Pierre Infelta et Michael Graetzel, Thermodynamique : Principes et Applications, BrownWalker Press, , 484 p. (ISBN 978-1-58112-995-3, lire en ligne), p. 118-120.
- Site Carnotcycle, Coefficient de Joule-Thomson(en).
- Jacques Schwartzentruber, École nationale supérieure des mines d'Albi-Carmaux, « Équation d'état analytique : interprétation des différentes branches », sur nte.mines-albi.fr, (consulté le ).
- L'eau dans tous ses états, Le temps, David Larousserie, 21 mai 2014.
- (en) Bharat Bhushan, Scanning Probe Microscopy in Nanoscience and Nanotechnology, vol. 2, Springer Science & Business Media, , 816 p. (ISBN 9783642104978, lire en ligne), p. 554-555.
- Joseph Kane, Morton Sternheim, Philippe Ghosez, Maryse Hoebeke et Gabriel Llabrés (trad. de l'anglais), Physique, Dunod, , 4e éd., 912 p. (lire en ligne), p. 390.
- (en) Jochen Vogt, Exam Survival Guide : Physical Chemistry, Springer, , 382 p. (ISBN 9783319498102, lire en ligne), p. 26-29.
- Weast. R. C. (Ed.), Handbook of Chemistry and Physics (53rd ed.), Cleveland: Chemical Rubber Co., 1972.





Bibliographie
- Article
- Daniel Berthelot, « Sur la notion des états correspondants et sur divers points correspondants remarquables », Journal de Physique Théorique et Appliquée, vol. 2, no 1, , p. 186–202 (ISSN 0368-3893, DOI 10.1051/jphystap:019030020018600, lire en ligne, consulté le ).
- Livres
- Christophe Coquelet et Dominique Richon, Propriétés thermodynamiques : Détermination pour les fluides purs, vol. BE 8030, Techniques de l'ingénieur, coll. « base documentaire Thermodynamique et énergétique, pack Physique énergétique, univers : Énergies », , p. 1-8.
- Jean-Pierre Corriou, Thermodynamique chimique : Diagrammes thermodynamiques, vol. J 1026, Techniques de l'ingénieur, coll. « base documentaire Thermodynamique et cinétique chimique, pack Opérations unitaires. Génie de la réaction chimique, univers Procédés chimie - bio - agro », , p. 1-30.
- Richard Taillet, Loïc Villain et Pascal Febvre, Dictionnaire de physique : + de 6500 termes, nombreuses références historiques, des milliers de références bibliographiques, Louvain-la-Neuve/impr. aux Pays-Bas, De Boeck supérieur, , 976 p. (ISBN 978-2-8073-0744-5, lire en ligne), p. 765-766.
- Jean Vidal, Thermodynamique : application au génie chimique et à l'industrie pétrolière, Paris, Editions Technip, coll. « Publications de l'Institut français du pétrole. », , 500 p. (ISBN 978-2-7108-0715-5, OCLC 300489419, lire en ligne).
- (en) Mark W. Zemansky et Richard H. Dittman, Heat and Thermodynamics : An Intermediate Textbook, McGraw-Hill, , 7e éd., 487 p. (ISBN 0-07-017059-2, lire en ligne [PDF]).
Liens externes
- Patrick Eggli, Laboratoires de Recherche et de Chimie Bienne (LRCB Sarl, « L’équation d’état de Van der Waals » [PDF], sur lrcb.ch, (consulté le ).
- Guy Collin, Université du Québec à Chicoutimi (UQAC), « Cours de chimie physique - Chapitre 3 - Le gaz réel », sur uqac.ca, (consulté le ).
- Éric Brunet, Jérôme Beugnon et Élie Wandersman, Laboratoire de Physique Statistique, École Normale Supérieure, « Cours de physique statistique » [PDF], sur lps.ens.fr, (consulté le ).
- Claude Rozé, Université de Rouen-Normandie, « Thermodynamique » [PDF], sur gsi-energie.univ-rouen.fr, (consulté le ).
- Patrick Puzo, Université Paris-Saclay, LAL, « Thermodynamique classique : Chapitre 6 - Description des fluides réels » [PDF], sur users.lal.in2p3.fr (consulté le ).
- Claude Saint-Blanquet, Université de Nantes, « Exercices de Thermodynamique sur les corps réels », sur sciences.univ-nantes.fr (consulté le ).
- Jean Bernard, Process's, « Effet Joule-Thomson », sur processs.free.fr (consulté le ).
Articles connexes
 Portail de la physique
Portail de la physique  Portail de la chimie
Portail de la chimie