Substance amyloïde
La substance amyloïde est un agrégat de protéines qui se plient sous une forme permettant à de nombreuses copies de cette protéine de s'agglutiner les unes aux autres et de constituer ainsi des fibrilles.
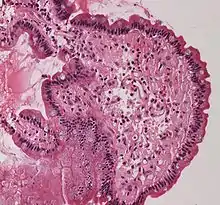
Dans le corps humain, la substance amyloïde est liée au développement de diverses maladies. Une telle substance amyloïde pathogène se forme lorsque des protéines précédemment saines perdent leurs fonctions physiologiques normales et s'agrègent en dépôts fibreux autour des cellules dans des plaques susceptibles de perturber le bon fonctionnement des tissus et des organes. Cette substance amyloïde a été associée (sans nécessairement en être la cause) à plus de 50 maladies humaines[1] connues sous le nom d'amyloïdoses, et peut jouer un rôle dans certaines maladies neurodégénératives[2].
Certaines protéines amyloïdes sont infectieuses et sont alors appelées prions[3] : la forme infectieuse peut servir de matrice pour convertir d'autres protéines non infectieuses en une forme infectieuse, car la forme inhabituelle du repliement et l'agrégation font alors partie des informations biologiques transmises[4].
La substance amyloïde peut aussi avoir des fonctions biologiques normales, par exemple, dans la formation de fimbriae chez certains genres de bactéries, la transmission de traits épigénétiques chez les champignons, ainsi que dans le dépôt de pigments et la libération d'hormones chez l'homme[5].
Cette substance amyloïde peut naître à partir de nombreuses protéines différentes[6]. Ces chaînes polypeptidiques forment généralement des structures en feuillets β qui s'agrègent en fibres longues ; cependant, des polypeptides identiques peuvent se replier en plusieurs conformations amyloïdes distinctes. La diversité des conformations peut avoir conduit à différentes formes de maladies à prions[5].
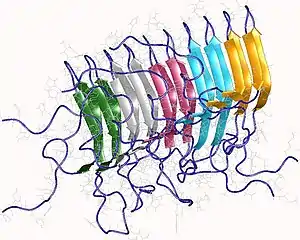
Définition
Le nom amyloïde provient de l'identification erronée de la substance par Rudolf Virchow qui a cru y reconnaître, à partir de techniques grossières de coloration à l'iode, une forme d'amidon (amylum en latin, du grec mistμυλον amylon). Pendant un certain temps, la communauté scientifique a débattu de la question de savoir si les dépôts amyloïdes étaient des dépôts graisseux ou glucidiques jusqu'à ce qu'il soit finalement découvert en 1859 qu'il s'agissait en réalité de dépôts de matière protéique albumoïde[7].
- La définition histopathologique classique de la substance amyloïde est celle de dépôts protéiques extracellulaires présentant une structure en feuillets bêta. Comme la plupart des structures de type bêta croisé, ils sont généralement identifiés par la biréfringence vert pomme lorsqu'ils sont colorés au rouge Congo et observés sous une lumière polarisée. Ces dépôts recrutent souvent divers sucres et autres composants tels que le sérum amyloïde S, ce qui entraîne des structures complexes et parfois non homogènes[8]. Récemment, cette définition a été remise en question, car certaines formes amyloïdes classiques ont été observées à l'intérieur de cellules[9].
- Une définition biophysique plus récente plus large inclut tout polypeptide qui se polymérise pour former une structure bêta-croisée, in vivo ou in vitro. Certains d'entre eux, bien qu'étant manifestement des feuillets bêta-croisés, ne présentent pas certaines caractéristiques histopathologiques classiques telles que la biréfringence rouge Congo[réf. nécessaire]. Les microbiologistes et biophysiciens ont largement adopté cette définition[10], [11], conduisant à certains conflits dans la communauté biologique sur une question de langage .
Le reste de cet article utilise la définition biophysique.
Substance amyloïdes pathogènes
Chez l'être humain et les autres mammifères
La société internationale des amyloidoses classe les fibrilles amyloïdes sur la base des protéines associées[28].
Substance amyloïde non pathologique et fonctionnelle
- Amyloïdes natifs dans les organismes [30]
- Fibrilles de curli produites par E. coli , Salmonella et quelques autres membres des Enterobacteriales (Csg). Les éléments génétiques (opérons) codant le système de curli sont largement répandus dans le système phylogénétique et peuvent être trouvés dans au moins quatre phyla bactériens[31]. Ceci suggère que beaucoup plus de bactéries peuvent exprimer des fibrilles de curli.
- Vésicules de gaz, les organites de flottabilité des archées aquatiques et des eubactéries [32]
- Amyloïdes fonctionnels dans Pseudomonas (Fap)[33],[34]
- Protéine d' enveloppe du paludisme
- Soie d'araignée (certaines araignées mais pas toutes)
- Mélanosomes mammaliennes (PMEL)
- Activateur de plasminogène de type tissulaire (tPA), facteur hémodynamique.
- Protéine ApCPEB et ses homologues avec un domaine riche en glutamine
- Les protéines d'adhésion des cellules fongiques s'agrégeant à la surface des champignons pour former des régions amyloïdes à la surface des cellules avec une force de liaison considérablement accrue [35],[36].
- Les chaplins [37] de la bactérie Streptomyces coelicolor[38].
- Prion het-s de Podospora anserina[29].
- Hormones peptidiques / protéiques stockées sous forme d'amyloïdes dans des granules de sécrétion endocriniennes[39].
- Protéines et peptides mis au point pour fabriquer de l'amyloïde possédant des propriétés spécifiques, tels que des ligands qui ciblent les récepteurs de la surface cellulaire[40].
- Les amyloïdes fonctionnels sont abondants dans la plupart des biofilms environnementaux en fonction de la coloration avec des colorants et des anticorps spécifiques à l'amyloïde[41].
- Les gaines tubulaires renfermant les filaments de Methanosaeta thermophila sont les premiers amyloïdes fonctionnels à avoir été rapportés dans le domaine de la vie archéale[42].
Les dépôts amyloïdes d'ATTR provenant de la transthyrétine se produisent non seulement dans l'amylose héréditaire liée à la transthyrétine, mais également dans les cas avancés de vieillissement dans de nombreux tissus, chez de nombreuses espèces de mammifères. Ils sont fréquemment rencontrés lors des autopsies de supercentenaires. Une proposition est qu'ils pourraient médier certaines pathologies tissulaires observées lors du vieillissement avancé et limiter la durée de la vie humaine [43].
« Des dépôts amyloïdes apparaissent dans le pancréas de patients atteints de diabète sucré, bien que l’on ignore si cela est important du point de vue fonctionnel. Le composant principal de l'amyloïde pancréatique est un peptide de 37 résidus d'acides aminés connu sous le nom de polypeptide amyloïde d'îlot ou amyline. Celui-ci est stocké avec de l'insuline dans des granules de sécrétion dans des cellules B et est co-sécrété avec de l'insuline. » Il retarde le vidage gastrique et inhibe la sécrétion d'insuline[44].
Caractéristiques biophysiques
Structure
Les amyloïdes sont constitués de longues fibres non ramifiées caractérisées par une structure quaternaire à feuillets bêta croisés dans laquelle des chaînes de peptides à brin β sont disposées perpendiculairement à l'axe de la fibre. Chaque fibre individuelle peut avoir une largeur de 5 à 15 nanomètres et une longueur de quelques micromètres[5]. Alors que l’amyloïde est généralement identifiée à l’aide de colorants fluorescents, de polarimétrie de coloration, de dichroïsme circulaire ou de mesures FTIR (mesures indirectes), le test "gold-standard" permettant de déterminer si une structure contient des fibres croisées consiste à placer un échantillon dans un faisceau de diffraction de rayons X. Le terme « β croisé » est basé sur l'observation de deux ensembles de raies de diffraction, l'une longitudinale et l'autre transversale, qui forment un motif « en croix » caractéristique[45]. Il existe deux signaux caractéristiques de diffraction par diffusion produits à 4,7 et 10 Angstroms (0,47 nm et 1,0 nm), correspondant aux distances d'interstrand et d'empilement en feuilles bêta[46]. Les "piles" de feuilles bêta sont courtes et traversent la largeur de la fibrille amyloïde; la longueur de la fibrille amyloïde est construite par des brins alignés. Le motif bêta-croisé est considéré comme une caractéristique diagnostique de la structure de l'amyloïde.
Pendant longtemps, notre connaissance de la structure au niveau atomique des fibrilles amyloïdes était limitée, les méthodes les plus traditionnelles d'étude des structures protéiques n'étant pas adaptées. Au cours des dernières années, des progrès ont été enregistrés dans les méthodes expérimentales, qui permettent désormais d’obtenir des données directes sur la structure interne de différents types de fibrilles amyloïdes. Deux méthodes bien connues incluent l’utilisation de la spectroscopie RMN à l’état solide et de la microscopie (cryo) électronique. Combinées, ces méthodes ont fourni des structures atomiques 3D de fibrilles amyloïdes formées par des peptides bêta-amyloïdes, de l'α-synucléine, des protéines tau et la protéine FUS, associées à diverses maladies neurodégénératives[47],[48].
Des études de diffraction des rayons X sur des microcristaux ont révélé des détails atomistiques de la région centrale de l'amyloïde [49],[50]. Les structures cristallographiques montrent que de courtes extensions à partir de régions de protéines amyloïdogènes à tendance amyloïde s'étendent perpendiculairement à l'axe du filament, ce qui correspond au caractère « β croisé » de la structure amyloïde. Elles révèlent également un certain nombre de caractéristiques des structures amyloïdes - les feuillets β voisins sont étroitement liés entre eux via une interface sans eau (appelée interface sèche), les brins β opposés étant légèrement décalés les uns des autres, de sorte que les chaînes s'imbriquent. Cette interface compacte déshydratée créée a été appelée interface stérique ou fermeture Éclair[5]. Il existe huit classes théoriques d'interfaces de type fermetures Éclair stériques, déclinées en fonction de la directionnalité des feuilles β (parallèle et anti-parallèle) et par la symétrie entre les feuillets β adjacents.
Bien que les structures amyloïdes authentiques soient toujours basées sur des feuillets β intermoléculaires, différents types de plis tertiaires "d'ordre supérieur" ont été observés ou proposés. Les feuillets β peuvent former un sandwich β, ou un solénoïde β qui peut être soit une hélice β, soit un rouleau β. Un facteur de complication dans les études de polypeptides amyloïdogènes est que des polypeptides identiques peuvent se replier en plusieurs conformations amyloïdes distinctes[5]. Ce phénomène est généralement décrit comme un polymorphisme amyloïde[51],[52],[53]. Ce polymorphisme se rencontre de façon omniprésente dans les substances amyloïdes pathogènes[54], et il pourrait expliquer la formation de souches de prions[réf. nécessaire].
Formation
La substance amyloïde est formée par la polymérisation de centaines à des milliers de peptides monomères en fibres longues. En général, la polymérisation amyloïde (agrégation ou polymérisation non covalente) est sensible à la séquence, c’est-à-dire que des mutations de la séquence peuvent empêcher l’auto-assemblage, en particulier si la mutation est un briseur de feuillet bêta, telle que la proline ou l'acide alpha-aminoisobutyrique non codé[55]. Par exemple, les humains produisent de l'amyline, un peptide amyloïdogène associé au diabète de type II, mais chez les rats et les souris, les prolines sont substituées dans des endroits critiques et aucune amyloïdogenèse ne se produit. [réf. nécessaire] Des études comparant l'amyloïde bêta 1-42 synthétique recombinant dans des dosages mesurant le taux de fibrillation, l'homogénéité des fibrilles et la toxicité cellulaire ont montré que l'amyloïde bêta 1-42 recombinant avait un taux de fibrillation plus rapide et une plus grande toxicité que l'amyloïde bêta synthétique 1-42 peptide[56]. Cette observation, combinée à l'irreproductibilité de certaines études expérimentales sur l'amyloïde bêta 1-42, pourrait être à l'origine du manque de progrès dans la recherche sur Alzheimer[57].
Il existe plusieurs classes de séquences polypeptidiques formant des amyloïdes[51],[52],[53]. Les polypeptides riches en glutamine jouent un rôle important dans l'amyloïdogenèse des prions de levure et de mammifère, ainsi que dans les troubles à répétition trinucléotidique, notamment la maladie de Huntington. Lorsque les polypeptides riches en glutamine ont une conformation en feuille β, les glutamines peuvent renforcer la structure en formant une liaison hydrogène entre les brins entre ses carbonyles amides et les azotes des chaînes principale et latérale. L'âge d'apparition de la maladie de Huntington montre une corrélation inverse avec la longueur de la séquence de la polyglutamine, avec des résultats analogues dans un système modèle de C. elegans avec des peptides de polyglutamine artificiels[58].
Parmi les résidus hydrophobes, les acides aminés aromatiques ont la plus forte propension amyloïdogène[59],[60].
Pour ces peptides, une polymérisation croisée (fibrilles d'une séquence polypeptidique entraînant la formation d'autres fibrilles d'une autre séquence) est observée in vitro et éventuellement in vivo. [réf. nécessaire] Ce phénomène est important, car il expliquerait la propagation inter-espèces de prions et les taux différentiels de propagation de prions, ainsi qu'un lien statistique entre la maladie d'Alzheimer et le diabète de type 2[61]. En général, plus la séquence peptidique est similaire, plus la polymérisation croisée est efficace, bien que des séquences totalement différentes puissent se polymériser de manière croisée et que des séquences très similaires puissent même être des "bloqueurs" qui empêchent la polymérisation. [réf. nécessaire] Les polypeptides ne polymériseront pas de façon croisée leurs homologues en miroir, suggérant que le phénomène implique des événements de liaison et de reconnaissance spécifiques. [réf. nécessaire]
Le processus d'agrégation rapide, les changements de conformation rapides ainsi que les effets de solvant constituent un défi pour la mesure des structures peptidiques amyloïdes monomères et oligomères en solution. Les études théoriques et informatiques complètent les expériences et fournissent des informations difficiles à obtenir avec des outils expérimentaux classiques. Plusieurs groupes ont étudié avec succès les structures désordonnées de substances amyloïdes et ont rapporté des structures de bobines aléatoires avec une structuration spécifique d'amyloïde monomère et oligomère, ainsi que l'impact de la génétique et du stress oxydatif sur les structures flexibles de la substance amyloïde en solution[62].
Les intermédiaires oligomères de l’insuline au cours de la fibrillation (plus toxiques que d’autres intermédiaires: natif, protofibrille et fibrilles) ont diminué la tension superficielle de la solution, ce qui indique des propriétés analogues à celles des détergents des oligomères et un rôle important des forces hydrophobes dans la cytotoxicité des oligomères[63].
Pathologie amyloïde
Les raisons de l'association entre maladie et substance amyloïde ne sont pas claires. Dans certains cas, les dépôts perturbent physiquement l'architecture des tissus, suggérant une perturbation de la fonction par un processus en masse. Un consensus émergent implique des intermédiaires préfibrillaires plutôt que des fibres amyloïdes matures dans la mort cellulaire[14],[64].
Une déréglement calcique a été observé dans des cellules exposées à des oligomères amyloïdes. Ces petits agrégats peuvent former des membranes bicouches lipidiques planaires à canaux ioniques. On a émis l’hypothèse selon laquelle la formation de canaux pourrait expliquer la dysrégulation calcique et le dysfonctionnement mitochondrial en permettant une fuite aveugle d’ions à travers les membranes cellulaires[65].
Des études ont montré que le dépôt d'amyloïde est associé à un dysfonctionnement mitochondrial et à la génération résultante d'espèces réactives de l'oxygène (ROS), qui peuvent initier une voie de signalisation conduisant à l'apoptose[66].
Des rapports indiquent que les polymères amyloïdes (tels que ceux de la huntingtine, associés à la maladie de Huntington) peuvent induire la polymérisation de protéines amyloïdogènes essentielles, qui devraient être nocives pour les cellules. En outre, des partenaires d'interaction de ces protéines essentielles peuvent également être séquestrés[67].
Coloration histologique
En milieu clinique, les maladies amyloïdes sont généralement identifiées par un changement d'intensité de fluorescence de colorants aromatiques plans tels que la thioflavine T, le rouge Congo ou le NIAD-4[68]. En général, cela est attribué au changement environnemental, car ces colorants s'intercalent entre des brins bêta pour confiner leur structure [69]. La positivité du rouge Congo reste la référence en matière de diagnostic de l’amylose. En général, la liaison du rouge Congo aux plaques amyloïdes produit une biréfringence vert pomme typique lorsqu'elle est observée sous une lumière polarisée croisée. Récemment, l'amélioration significative du rendement quantique de fluorescence de NIAD-4 a été exploitée pour l'imagerie par fluorescence à super-résolution de fibrilles amyloïdes[70] et d'oligomères[71]. Pour éviter une coloration non spécifique, d'autres colorations histologiques, telles que la coloration à l'hématoxyline et à l'éosine, sont utilisées pour éteindre l'activité des colorants à d'autres endroits, tels que le noyau, où le colorant pourrait se lier. La technologie moderne des anticorps et l'immunohistochimie ont facilité des colorations spécifiques, mais cela peut souvent poser problème car les épitopes peuvent être dissimulés dans le repli amyloïde. En général, une structure de protéine amyloïde a une conformation différente de celle reconnue par l'anticorps.
Voir également
- Protéopathie
- JUNQ et IPOD
- Hypothèse amyloïde
Références
- « The amyloid state and its association with protein misfolding diseases », Nature Reviews. Molecular Cell Biology, vol. 15, no 6, , p. 384–96 (PMID 24854788, DOI 10.1038/nrm3810)
- Tuomas P. J. Knowles, Michele Vendruscolo et Christopher M. Dobson, « Ubiquitous amyloids », Applied Biochemistry and Biotechnology, vol. 166, no 7, , p. 1626–43 (PMID 22350870, PMCID 3324686, DOI 10.1007/s12010-012-9549-3)
- Note: Certains auteurs font toutefois une distinction entre les prions et substances amyloïdes analogues aux prions (« prion-like »)
- Claudio Soto, Lisbell Estrada et Joaquín Castilla, « Amyloids, prions and the inherent infectious nature of misfolded protein aggregates », Trends in Biochemical Sciences, vol. 31, no 3, , p. 150–5 (PMID 16473510, DOI 10.1016/j.tibs.2006.01.002)
- « Amyloid structure: conformational diversity and consequences », Annual Review of Biochemistry, vol. 80, , p. 557–85 (PMID 21456964, PMCID 3817101, DOI 10.1146/annurev-biochem-090908-120656)
- « A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro », Proceedings of the National Academy of Sciences of the United States of America, vol. 97, no 16, , p. 8979–84 (PMID 10908649, PMCID 16807, DOI 10.1073/pnas.150091797)
- « Amyloidosis: a convoluted story », British Journal of Haematology, vol. 114, no 3, , p. 529–38 (PMID 11552976, DOI 10.1046/j.1365-2141.2001.02999.x)
- « Review: history of the amyloid fibril », Journal of Structural Biology, vol. 130, nos 2–3, , p. 88–98 (PMID 10940217, DOI 10.1006/jsbi.2000.4221)
- « Toxic human islet amyloid polypeptide (h-IAPP) oligomers are intracellular, and vaccination to induce anti-toxic oligomer antibodies does not prevent h-IAPP-induced beta-cell apoptosis in h-IAPP transgenic mice », Diabetes, vol. 56, no 5, , p. 1324–32 (PMID 17353506, DOI 10.2337/db06-1579)
- « Techniques to study amyloid fibril formation in vitro », Methods, vol. 34, no 1, , p. 151–60 (PMID 15283924, DOI 10.1016/j.ymeth.2004.03.012)
- « On the structural definition of amyloid fibrils and other polypeptide aggregates », Cellular and Molecular Life Sciences, vol. 64, no 16, , p. 2066–78 (PMID 17530168, DOI 10.1007/s00018-007-7110-2)
- « The many faces of amyloid beta in Alzheimer's disease », Current Molecular Medicine, vol. 8, no 6, , p. 580–4 (PMID 18781964, DOI 10.2174/156652408785747951)
- « Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases », Molecular Medicine, vol. 14, nos 7–8, , p. 451–64 (PMID 18368143, PMCID 2274891, DOI 10.2119/2007-00100.Irvine)
- « Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases », IUBMB Life, vol. 59, nos 4–5, , p. 332–45 (PMID 17505973, DOI 10.1080/15216540701283882)
- « The amyloid beta peptide: a chemist's perspective. Role in Alzheimer's and fibrillization », Chemical Reviews, vol. 112, no 10, , p. 5147–92 (PMID 22813427, DOI 10.1021/cr3000994, lire en ligne)
- « Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis », Endocrine Reviews, vol. 29, no 3, , p. 303–16 (PMID 18314421, PMCID 2528855, DOI 10.1210/er.2007-0037)
- « Islet amyloid and type 2 diabetes mellitus », The New England Journal of Medicine, vol. 343, no 6, , p. 411–9 (PMID 10933741, DOI 10.1056/NEJM200008103430607)
- « More than just mad cow disease », Nature Structural Biology, vol. 8, no 4, , p. 281 (PMID 11276238, DOI 10.1038/86132)
- Monari, L.1., Chen, S.G., Brown, P., Parchi, P., Petersen, R.B., Mikol, J., Gray, F., Cortelli, P., Montagna, P., Ghetti, B., « Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. », Proceedings of the National Academy of Science., vol. 91(7), , p. 2839-42
- « Huntington's disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases », The FEBS Journal, vol. 275, no 17, , p. 4252–62 (PMID 18637947, DOI 10.1111/j.1742-4658.2008.06561.x)
- « Targeting protein aggregation in neurodegeneration--lessons from polyglutamine disorders », Expert Opinion on Therapeutic Targets, vol. 10, no 4, , p. 505–13 (PMID 16848688, DOI 10.1517/14728222.10.4.505)
- Mann DM, Snowden JS, « Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype », Brain Pathology, vol. 27, no 6, , p. 723–736 (PMID 28100023, DOI 10.1111/bpa.12486)
- (en) Nelson, Dickson, Trojanowski et Boyle, « Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report », Brain, vol. Online first, (DOI 10.1093/brain/awz099, lire en ligne)
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL, « Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view », Journal of Neuropathology and Experimental Neurology, vol. 62, no 9, , p. 885–98 (PMID 14533778, DOI 10.1093/jnen/62.9.885
 )
) - « Amyloidosis: Definition of Amyloid and Amyloidosis, Classification Systems, Systemic Amyloidoses », sur eMedicine,
- « Alzheimer's dementia by circulation disorders: when trees hide the forest », Nature Cell Biology, vol. 11, no 2, , p. 114–6 (PMID 19188916, DOI 10.1038/ncb0209-114)
- « Amyloid fibril protein nomenclature: 2010 recommendations from the nomenclature committee of the International Society of Amyloidosis », Amyloid, vol. 17, nos 3–4, , p. 101–4 (PMID 21039326, DOI 10.3109/13506129.2010.526812)
- Reed B. Wickner, Herman K. Edskes, David A. Bateman et Amy C. Kelly, « Amyloids and yeast prion biology », Biochemistry, vol. 52, no 9, , p. 1514–1527 (ISSN 0006-2960, PMID 23379365, PMCID 7321840, DOI 10.1021/bi301686a, lire en ligne, consulté le )
- « Amyloids: friend or foe? », Journal of Alzheimer's Disease, vol. 13, no 4, , p. 407–19 (PMID 18487849, PMCID 2674399, DOI 10.3233/JAD-2008-13406, lire en ligne [archive du ])
- Mark Alexander Webber (dir.), « Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure », PLOS ONE, vol. 7, no 12, (PMID 23251478, PMCID 3521004, DOI 10.1371/journal.pone.0051274)
- « An amyloid organelle, solid-state NMR evidence for cross-β assembly of gas vesicles », The Journal of Biological Chemistry, vol. 287, no 5, , p. 3479–84 (PMID 22147705, PMCID 3271001, DOI 10.1074/jbc.M111.313049)
- « Functional amyloid in Pseudomonas », Molecular Microbiology, vol. 77, no 4, , p. 1009–20 (PMID 20572935, DOI 10.1111/j.1365-2958.2010.07269.x)
- « Expression of Fap amyloids in Pseudomonas aeruginosa, P. fluorescens, and P. putida results in aggregation and increased biofilm formation », MicrobiologyOpen, vol. 2, no 3, , p. 365–82 (PMID 23504942, PMCID 3684753, DOI 10.1002/mbo3.81)
- « A role for amyloid in cell aggregation and biofilm formation », PLOS ONE, vol. 6, no 3, , e17632 (PMID 21408122, PMCID 3050909, DOI 10.1371/journal.pone.0017632)
- « Strengthening relationships: amyloids create adhesion nanodomains in yeasts », Trends in Microbiology, vol. 20, no 2, , p. 59–65 (PMID 22099004, PMCID 3278544, DOI 10.1016/j.tim.2011.10.002)
- Les chaplins sont des agrégats de protéines de surface hydrophobes qui recouvrent le mycelium aérien - voir aussi The chaplins: a family of hydrophobic cell-surface proteins involved in aerial mycelium formation in Streptomyces coelicolor
- (en) Elizabeth B. Sawyer, Dennis Claessen, Maria Haas et Sally L. Gras, « The Assembly of Individual Chaplin Peptides from Streptomyces coelicolor into Functional Amyloid Fibrils », sur PLoS ONE, (DOI https://doi.org/10.1371/journal.pone.0018839).
- « Functional amyloids as natural storage of peptide hormones in pituitary secretory granules », Science, vol. 325, no 5938, , p. 328–32 (PMID 19541956, PMCID 2865899, DOI 10.1126/science.1173155)
- « Functional fibrils derived from the peptide TTR1-cycloRGDfK that target cell adhesion and spreading », Biomaterials, vol. 32, no 26, , p. 6099–110 (PMID 21636126, DOI 10.1016/j.biomaterials.2011.05.021)
- « Amyloid adhesins are abundant in natural biofilms », Environmental Microbiology, vol. 9, no 12, , p. 3077–90 (PMID 17991035, DOI 10.1111/j.1462-2920.2007.01418.x)
- « The Tubular Sheaths Encasing Methanosaeta thermophila Filaments Are Functional Amyloids », The Journal of Biological Chemistry, vol. 290, no 33, , p. 20590–600 (PMID 26109065, PMCID 4536462, DOI 10.1074/jbc.M115.654780)
- Coles et Young, « Supercentenarians and transthyretin amyloidosis: The next frontier of human life extension », Preventive Medicine, vol. 54, , S9–S11 (PMID 22579241, DOI 10.1016/j.ypmed.2012.03.003)
- Humphrey P. Rang, James M. Ritter, Rod J. Flower et Graeme Henderson, Rang & Dale's Pharmacology, Elsevier Health Sciences, , 8e éd., 776 p. (ISBN 978-0-7020-5497-6, lire en ligne), p. 385/
- Wormell RL. New fibres from proteins. Academic Press, 1954, p. 106.
- « Common core structure of amyloid fibrils by synchrotron X-ray diffraction », Journal of Molecular Biology, vol. 273, no 3, , p. 729–39 (PMID 9356260, DOI 10.1006/jmbi.1997.1348)
- « Emerging Structural Understanding of Amyloid Fibrils by Solid-State NMR », Trends in Biochemical Sciences, vol. 42, no 10, , p. 777–787 (PMID 28916413, DOI 10.1016/j.tibs.2017.08.001)
- « Cryo-EM structures of tau filaments from Alzheimer's disease », Nature, vol. 547, no 7662, , p. 185–190 (PMID 28678775, PMCID 5552202, DOI 10.1038/nature23002)
- « Structure of the cross-beta spine of amyloid-like fibrils », Nature, vol. 435, no 7043, , p. 773–8 (PMID 15944695, PMCID 1479801, DOI 10.1038/nature03680)
- « Atomic structures of amyloid cross-beta spines reveal varied steric zippers », Nature, vol. 447, no 7143, , p. 453–7 (PMID 17468747, DOI 10.1038/nature05695)
- « Amyloid fibril formation by A beta 16-22, a seven-residue fragment of the Alzheimer's beta-amyloid peptide, and structural characterization by solid state NMR », Biochemistry, vol. 39, no 45, , p. 13748-59 (PMID 11076514, DOI 10.1021/bi0011330, lire en ligne)
- « Molecular alignment within beta-sheets in Abeta(14-23) fibrils: solid-state NMR experiments and theoretical predictions », Biophysical Journal, vol. 92, no 2, , p. 594-602 (PMID 17056725, PMCID 1751388, DOI 10.1529/biophysj.106.091017, lire en ligne)
- « Assembling amyloid fibrils from designed structures containing a significant amyloid beta-peptide fragment », Biochemical Journal, vol. 366, , p. 343-351 (PMID 12023906, PMCID 1222771, DOI 10.1042/BJ20020229, lire en ligne)
- Kevin C. Stein et Heather L. True, « Prion Strains and Amyloid Polymorphism Influence Phenotypic Variation », sur Plos, (DOI 10.1371/journal.ppat.1004328).
- « Inhibition of amyloid fibril formation by peptide analogues modified with alpha-aminoisobutyric acid », Angewandte Chemie, vol. 43, no 31, , p. 4041–4 (PMID 15300690, DOI 10.1002/anie.200353565)
- « The recombinant amyloid-beta peptide Abeta1-42 aggregates faster and is more neurotoxic than synthetic Abeta1-42 », Journal of Molecular Biology, vol. 396, no 1, , p. 9–18 (PMID 20026079, DOI 10.1016/j.jmb.2009.12.016)
- « State of aggregation », Nature Neuroscience, vol. 14, no 4, , p. 399 (PMID 21445061, DOI 10.1038/nn0411-399)
- « The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans », Proceedings of the National Academy of Sciences of the United States of America, vol. 99, no 16, , p. 10417–22 (PMID 12122205, PMCID 124929, DOI 10.1073/pnas.152161099)
- « A possible role for pi-stacking in the self-assembly of amyloid fibrils », FASEB Journal, vol. 16, no 1, , p. 77–83 (PMID 11772939, DOI 10.1096/fj.01-0442hyp)
- « Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases », Journal of Molecular Biology, vol. 350, no 2, , p. 379–92 (PMID 15925383, DOI 10.1016/j.jmb.2005.04.016)
- « Amylin deposition in the brain: A second amyloid in Alzheimer disease? », Annals of Neurology, vol. 74, no 4, , p. 517–26 (PMID 23794448, PMCID 3818462, DOI 10.1002/ana.23956)
- « Amyloid-β peptide structure in aqueous solution varies with fragment size », The Journal of Chemical Physics, vol. 135, no 20, , p. 205101 (PMID 22128957, DOI 10.1063/1.3662490)
- « Oligomeric forms of insulin amyloid aggregation disrupt outgrowth and complexity of neuron-like PC12 cells », PLOS ONE, vol. 7, no 7, , e41344 (PMID 22848469, PMCID 3407202, DOI 10.1371/journal.pone.0041344)
- « Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers », The Journal of Biological Chemistry, vol. 280, no 17, , p. 17294–300 (PMID 15722360, DOI 10.1074/jbc.M500997200)
- « Amyloid peptide channels », The Journal of Membrane Biology, vol. 202, no 1, , p. 1–10 (PMID 15702375, DOI 10.1007/s00232-004-0709-4)
- « Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation », Cell Death and Differentiation, vol. 12, no 1, , p. 19–24 (PMID 15592360, DOI 10.1038/sj.cdd.4401528)
- Tuite (dir.), « Amyloid-mediated sequestration of essential proteins contributes to mutant huntingtin toxicity in yeast », PLOS ONE, vol. 7, no 1, , e29832 (PMID 22253794, PMCID 3256205, DOI 10.1371/journal.pone.0029832)
- « In vivo optical imaging of amyloid aggregates in brain: design of fluorescent markers », Angewandte Chemie, vol. 44, no 34, , p. 5452–6 (PMID 16059955, DOI 10.1002/anie.200500845)
- « Torsion-dependent fluorescence switching of amyloid-binding dye NIAD-4 », Chemical Physics Letters, vol. 633, , p. 109–13 (DOI 10.1016/j.cplett.2015.05.010)
- « Superresolution imaging of amyloid fibrils with binding-activated probes », ACS Chemical Neuroscience, vol. 4, no 7, , p. 1057–61 (PMID 23594172, PMCID 3715833, DOI 10.1021/cn400091m)
- « Morphological analysis of oligomeric vs. fibrillar forms of α-synuclein aggregates with super-resolution BALM imaging », Chemical Physics Letters, vol. 690, , p. 62–67 (DOI 10.1016/j.cplett.2017.10.034)
Liens externes
 Portail de la biochimie
Portail de la biochimie  Portail de la médecine
Portail de la médecine