Monomelic amyotrophy
| Monomelic amyotrophy | |
|---|---|
| Other names: Juvenile muscular atrophy of distal upper extremity, Hirayama disease, spinal muscular atrophy juvenile nonprogressive | |
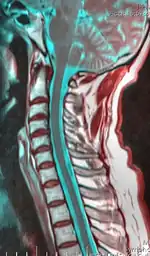 | |
| Cervical spine | |
Monomelic amyotrophy (MMA), is a rare motor neuron disease first described in 1959 in Japan. Its symptoms usually appear about two years after adolescent growth spurt and is significantly more common in males (average age of onset, 15- to 25-year-old). MMA is reported most frequently in Asia but has a global distribution. It is typically marked by insidious onset of muscle atrophy of an upper limb, which plateaus after two to five years from which it neither improves nor worsens. There is no pain or sensory loss associated with MMA. MMA is not believed to be hereditary.[1][2][3][4]
Both the names for the disorder and its possible causes have been evolving since first reported in 1959. It is most commonly believed the condition occurs by asymmetrical compression of the cervical spinal column by the cervical dural sac, especially when the neck is flexed. However, the disease is uncommon and diagnosis is confused by several atypical reports.[5][6][7]
Symptoms and signs

In terms of the signs and symptoms that are consistent for an individual who has monomelic amyotrophy are the following (although this does not reflect a complete list):[1]
- Muscle weakness
- Fasciculations
- Tremor
- Cold hands
- Muscle cramps
Initially most people notice weakness in one hand; they may feel contracture of middle and ring finger and notice a thinning of the subdigital palm of the affected fingers. Progress of the condition varies, and weakness in the arm ranges from minimal to significant. Fasciculations are uncommon (>20%); increased weakness under cold conditions is commonly reported (cold paresis).
Cause
The disability originates with impaired functioning of the anterior horn cells of the lower cervical cord (lower neck), but the cause of the decline is not fully understood and is still considered unknown.[8] Researchers, including Hirayama, believe that "forward displacement of the cervical dural sac and compressive flattening of the lower cervical cord during neck flexion"[9] is the contributing factor. Studies consistently note a loss of normal neck curvature (the cervical lordosis) and compression of the cervical chord by the dural sac in forward flexion.[10][11]
- "There is a debate about whether this condition represents a focal form of primary LMN degeneration (ie, a focal form of spinal muscular atrophy) or a local consequence of chronic compression from a dural expansion in the cervical spine."[6]
A familial link has been found in a minor percentage of cases, including parent-child and sibling-sibling. Because of the unusual distribution of the disease, some researchers speculate that there could be an ethnic link.[9][12][13]
Diagnosis
The condition presents almost exclusively in 15 to 25 year old adults experiencing weakness in hand and arm. A patient history and a neurological exam narrows down the possible diagnosis; this preliminary exam typically includes strength and reflex tests. Cold paresis (increased weakness in cold weather) is reported by most patients (> 80%). Fasciculations are reported as uncommon (< 20%) to common; larger tremors are more consistently cited. Males are far more likely to be diagnosed with the condition.[lower-alpha 1][14] [15]
The disease is rare and several cited cases deviate from the expected norm, making diagnosis more difficult. Proposed diagnostic criteria:
- Distal predominant muscle weakness and atrophy in forearm and hand
- Involvement of the unilateral upper extremity almost always all the time
- Onset between the ages of 10 to early 20s
- Insidious onset with gradual progression for the first several years, followed by stabilization
- No lower extremity involvement
- No sensory disturbance and tendon reflex abnormalities
- Exclusion of other diseases (e.g., motor neuron disease, multifocal motor neuropathy, brachial plexopathy, spinal cord tumors, syringomyelia, cervical vertebral abnormalities, anterior interosseous, or deep ulnar neuropathy)[5]
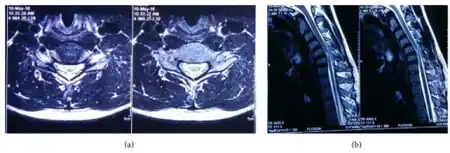
A neurological exam can suggest different motor neuron diseases (such as MMA), but to more confidently distinguish MMA from the diseases it mimics, advanced diagnostic tools are called for. These include exam tools such as Magnetic resonance imaging (MRI), and electromyography (EMG) and nerve conduction velocity (NCV) tests. An MRI examination of the neck would typically reveal - for a positive MMA diagnosis—some constriction of the cervical cord and an abnormal forward extension of the neck, ("loss of cervical lordosis"); pressure by the dura on the nerve cord apparently causes the flattening / narrowing. An EMG test reveals loss of the nerve supply, or denervation, in the affected limb without a conduction block (a nerve blockage restricted to a small segment of the nerve).
In early stages of the disease MMA may be confused for amyotrophic lateral sclerosis (ALS), cervical spondylotic amyotrophy (CSA), and other challenging neurological diseases, as well as conditions that are minor but that call for very different treatments, such as advanced carpal tunnel syndrome (CTS). Symptoms somewhat differ.[15][3] Pain and tingling in the hand is typically present in CTS and absent from MMA; loss of function presents differently; with careful electrophysiological study and neurological exams the two are distinguished. In early stages, ALS, SCA, and MMA, presentation may be similar. Both CSA and ALS ultimately have more extensive symptoms. MMA is more prevalent in young people while ALS and CSA are more common in older populations. With ALS, hand symptoms usually more commonly both proximal and distal vs in MMA mostly distal only, and with ALS fasciculations (twitching) are often present in upper extremities, but rarely in MMA. MMA is usually eliminated from consideration if disability expresses itself in more than one extremity or in lower extremities (legs), but symptomatic absence may not rule out ALS for three to five years after initial onset. Electrophysiological texts and reflex tests tend to yield different results, but interpretation is at times subjective.[3][16]
Treatment
At present there is no cure for MMA. The impact on the affected individual ranges from minimal to significant depending on the extent of the weakness. Physical and occupational therapies include muscle strengthening exercises and training in hand coordination.[12] Early use of a cervical collar is increasingly encouraged as therapeutic for arresting further compression of the cervical spinal cord.[11] Spinal surgery on patients with more advanced symptoms has met with reported success, but is still regarded as experimental.[17][18]
Prognosis
The symptoms of MMA usually progress slowly for two to five years and then remain stable for many years. The weakness can progress to the opposite limb, although whether this progression is typical or rare is under discussion. Cases of patient improvement and deterioration have both been described, but are atypical. Initially MMA can be confused for slowly progressing case of other neurological diseases such as amyotrophic lateral sclerosis (ALS); initial symptoms can be similar, but their causes are apparently different, and their outcomes markedly so. Diagnostic tools have improved since first described, and a few therapies are being introduced. But sometimes several years of observation are needed before a definitive diagnosis can be made.
Use of a cervical collar may afford relief, and some researchers advocate its therapeutic use. There is also a slowly progressive variant of MMA known as O'Sullivan-McLeod syndrome, which only affects the small muscles of the hand and forearm and has a slowly progressive course.[19]
Epidemiology
MMA is described most frequently in Asia, with studies of a few hundred individuals emerging from Japan, China and India; it is much less commonly seen in North America and Europe. The disease (disorder) was first described by Keizo Hirayama in 1959 as "juvenile muscular atrophy of unilateral upper extremity". In 1984 Mandavilli Gourie-Devi (et al) introduced the term “monomelic amyotrophy”. The disease primarily (but not exclusively) affects young (15- to 25-year-old) males. As of 2014 there had been less than 1500 described cases, starting with the patients in Hirayama's 1959 study. The condition is disproportionately high in Asia but no conclusive reason has been found for this. To date the largest studies recorded are Japan (333 cases),[20] India (279 cases),[10] and China (179 cases).[21]
Etymology
Both the names for the disorder and its possible causes have been evolving since first reported.[6] Because this condition has been found almost exclusively in healthy young adults and stabilizes after a few years, a span of 23 years elapsed between the time Hirayama first described the condition, and the first death (from cancer), and autopsy. Over the several decades since first reported, a majority of researchers now describe this as a biomechanical condition linked to adolescent growth spurt. However, this is not yet a universal conclusion and there are anomalous reports that suggest an incomplete understanding of the condition.[22]
See also
Notes
- ↑ The male to female ratio most commonly given is 10:1, but varies markedly in studies, from 5:2 (Bademkiran et al 2015) to 20:1 (Jin et al 2014)
References
- 1 2 "Monomelic amyotrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 24 December 2016. Retrieved 23 December 2016.
- ↑ "Monomelic Amyotrophy Information Page | National Institute of Neurological Disorders and Stroke". www.ninds.nih.gov. Archived from the original on 9 April 2020. Retrieved 23 December 2016.
- 1 2 3 Ay, Halil (22 May 2017). "Hirayama disease (monomelic amyotrophy) clinically confused for carpal tunnel syndrome". Neuropsychiatric Disease and Treatment. 2017:13: 1385–1388. doi:10.2147/NDT.S138315. PMC 5449119. PMID 28579784.
- ↑ Liewluck, Teerin; Saperstein, David S (November 2015). "Progressive Muscular Atrophy". In Dimachkie, Mazen M.; Barohn, Richard J. (eds.). Motor Neuron Disease, An Issue of Neurologic Clinics. Elsevier Health Sciences. p. 766. ISBN 9780323413459. Archived from the original on 2017-04-09. Retrieved 2021-08-28.
- 1 2 Anuradha, S; Fanai, V (2016). "Hirayama Disease: A Rare Disease with Unusual Features". Case Reports in Neurological Medicine. 2016: 5839761. doi:10.1155/2016/5839761. PMC 5209606. PMID 28097028.
- 1 2 3 Turner, Martin R; Talbot, Kevin (June 2013). "Mimics and chameleons in motor neurone disease". Practical Neurology. 13 (3): 153–164. doi:10.1136/practneurol-2013-000557. PMC 3664389. PMID 23616620.
- ↑ Hassan, KM; Sahni, H; Jha, A (April 2012). "Clinical and radiological profile of Hirayama disease: A flexion myelopathy due to tight cervical dural canal amenable to collar therapy". Annals of Indian Academy of Neurology. 15 (2): 106–112. doi:10.4103/0972-2327.94993. PMC 3345586. PMID 22566723.
- ↑ Polo, A; Curro' Dossi, M; Fiaschi, A; Zanette, GP; Rizzuto, N (May 2003). "Peripheral and segmental spinal abnormalities of median and ulnar somatosensory evoked potentials in Hirayama's disease". Journal of Neurology, Neurosurgery, and Psychiatry. 74 (5): 627–32. doi:10.1136/jnnp.74.5.627. PMC 1738443. PMID 12700306.
- 1 2 Hirayama, K.; Tokumaru, Y. (23 May 2000). "Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity". Neurology. 54 (10): 1922–1926. doi:10.1212/WNL.54.10.1922. PMID 10822430. S2CID 35938208.
- 1 2 Nalini, A; Gourie-Devi, M; Thennarasu, K; Ramalingaiah, AH (September 2014). "Monomelic amyotrophy: clinical profile and natural history of 279 cases seen over 35 years (1976-2010)". Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration. 15 (5–6): 457–65. doi:10.3109/21678421.2014.903976. PMID 24853410. S2CID 34932891. (179 cases)
- 1 2 Hassan KM, Sahni H (2013). "Nosology of juvenile muscular atrophy of distal upper extremity: from monomelic amyotrophy to Hirayama disease--Indian perspective". Biomed Res Int. 2013: 478516. doi:10.1155/2013/478516. PMC 3770029. PMID 24063005.
- 1 2 "OMIM Entry - % 602440 - AMYOTROPHY, MONOMELIC". www.omim.org. Archived from the original on 8 July 2020. Retrieved 24 May 2018.
- ↑ Gücüyener, Kivilcim; Aysun, Sabiha; Topaloglu, Haluk; Inan, Levent; Varli, Kubilay; Griffith, BP (May 1991). "Monomelic amyotrophy in siblings". Pediatric Neurology. 7 (3): 220–222. doi:10.1016/0887-8994(91)90089-4. PMID 1878104.
- ↑ Bademkiran, Fikret; Oto, Aycan; Tabakoglu, Ayçin; Aydogdu, I; Uludağ, Burhanettin (2015). "Monomelic Amyotrophy (Hirayama Disease): Clinical Findings, EMG Characteristics and Differential Diagnosis" (PDF). Journal of the Neurological Sciences (in Türkçe). 32 (3:45): 558–565. Archived from the original (PDF) on 2018-09-14. Retrieved 2021-08-28.
- 1 2 Jin, X; Jiang, JY; Lu, FZ; Xia, XL; Wang, LX; Zheng, CJ (16 October 2014). "Electrophysiological differences between Hirayama disease, amyotrophic lateral sclerosis and cervical spondylotic amyotrophy". BMC Musculoskeletal Disorders. 15: 349. doi:10.1186/1471-2474-15-349. PMC 4216382. PMID 25319248.
- ↑ Talbot, Kevin (1 December 2004). "Monmelic Amyotrophy Hirayama's Disease". Practical Neurology. 4 (6): 362–365. doi:10.1111/j.1474-7766.2004.00265.x.
- ↑ Lin MS, Kung WM, Chiu WT, Lyu RK, Chen CJ, Chen TY (June 2010). "Hirayama disease". J Neurosurg Spine. 12 (6): 629–34. doi:10.3171/2009.12.SPINE09431. PMID 20515348.
- ↑ Paredes, I; Esteban, J; Ramos, A; Gonzalez, P; Rivas, JJ (February 2014). "A severe case of Hirayama disease successfully treated by anterior cervical fusion". Journal of Neurosurgery: Spine. 20 (2): 191–5. doi:10.3171/2013.10.SPINE13508. PMID 24286527. S2CID 41987942. Archived from the original on 2021-09-23. Retrieved 2021-08-28.
- ↑ Ghadiri-Sani, M; Huda, S; Larner, AJ (December 2014). "O'Sullivan-McLeod syndrome: clinical features, neuroradiology and nosology". British Journal of Hospital Medicine. 75 (12): 712–3. doi:10.12968/hmed.2014.75.12.712. PMID 25488537.
- ↑ Hirayama, K (January 2008). "[Juvenile muscular atrophy of unilateral upper extremity (Hirayama disease)--half-century progress and establishment since its discovery]". Brain and Nerve = Shinkei Kenkyu No Shinpo. 60 (1): 17–29. PMID 18232329.
- ↑ Zhou, Bo; Chen, Lei; Fan, Dongsheng; Zhou, Dong (26 February 2010). "Clinical features of Hirayama disease in mainland China". Amyotrophic Lateral Sclerosis. 11 (1–2): 133–139. doi:10.3109/17482960902912407. PMID 19412815. S2CID 13040071.
- ↑ Lehman, V.T.; Luetmer, P.H.; Sorenson, E.J.; Carter, R.E.; Gupta, V.; Fletcher, G.P.; Hu, L.S.; Kotsenas, A.L. (February 2013). "Cervical Spine MR Imaging Findings of Patients with Hirayama Disease in North America: A Multisite Study". American Journal of Neuroradiology. 34 (2): 451–456. doi:10.3174/ajnr.A3277. PMC 7965093. PMID 22878010.
External links
| Classification | |
|---|---|
| External resources |
|