Immunomodulatory imide drug
| Immunomodulatory imide drug | |
|---|---|
| Drug class | |
 Thalidomide | |
| Class identifiers | |
| Use | Erythema nodosum leprosum, multiple myeloma, myelodysplastic syndrome, acute myeloid leukaemia and other immunologic conditions |
| ATC code | L04AX |
| Biological target | TNF, IL-6, VEGF, NF-kB, etc. |
| Clinical data | |
| Drugs.com | Drug Classes |
| In Wikidata | |
Immunomodulatory imide drugs (IMiDs) are a class of immunomodulatory drugs[1] (drugs that adjust immune responses) containing an imide group. The IMiD class includes thalidomide and its analogues (lenalidomide, pomalidomide, and iberdomide.[1] These drugs may also be referred to as 'Cereblon modulators'. Cereblon (CRBN) is the protein targeted by this class of drugs.
The name "IMiD" alludes to both "IMD" for "immunomodulatory drug" and the forms imide, imido-, imid-, and imid.
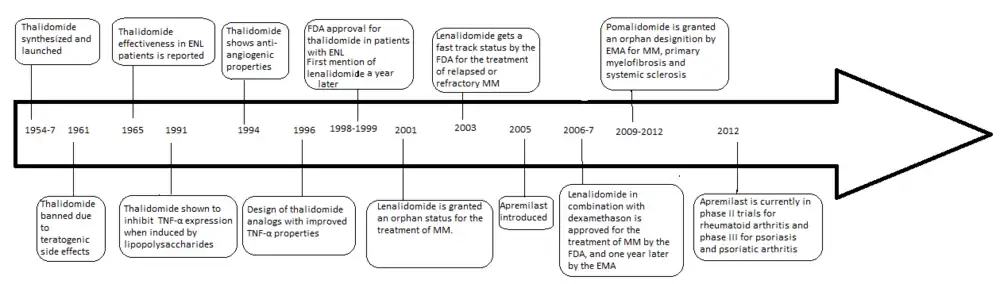
The development of analogs of thalidomide was precipitated by the discovery of the anti-angiogenic and anti-inflammatory properties of the drug yielding a new way of fighting cancer as well as some inflammatory diseases after it had been banned in 1961. The problems with thalidomide included; teratogenic side effects, high incidence of other adverse reactions, poor solubility in water and poor absorption from the intestines.
In 1998 thalidomide was approved by the U.S. Food and Drug Administration (FDA) for use in newly diagnosed multiple myeloma (MM) under strict regulations.[2] This has led to the development of a number of analogs with fewer side effects and increased potency which include lenalidomide and pomalidomide, which are currently marketed and manufactured by Celgene.
History
Thalidomide was originally released in the Federal Republic of Germany (West Germany) under the label of Contergan on October 1, 1957, by Chemie Grünenthal (now Grünenthal). The drug was primarily prescribed as a sedative or hypnotic, but it was also used as an antiemetic for morning sickness in pregnant women. The drug was banned in 1961 after its teratogenic properties were observed. The problems with thalidomide were, aside from the teratogenic side effects, both high incidence of other adverse reactions along with poor solubility in water and absorption from the intestines.[3][4] Adverse reactions include peripheral neuropathy in large majority of patients, constipation, thromboembolism along with dermatological complications.[5]
Four years after thalidomide was withdrawn from the market for its ability to induce severe birth defects, its anti-inflammatory properties were discovered when patients suffering from erythema nodosum leprosum (ENL) used thalidomide as a sedative and it reduced both the clinical signs and symptoms of the disease. Thalidomide was discovered to inhibit tumour necrosis factor-alpha (TNF-α) in 1991 (5a Sampaio, Sarno, Galilly Cohn and Kaplan, JEM 173 (3) 699–703, 1991) . TNF-α is a cytokine produced by macrophages of the immune system, and also a mediator of inflammatory response. Thus the drug is effective against some inflammatory diseases such as ENL (6a Sampaio, Kaplan, Miranda, Nery..... JID 168 (2) 408-414 2008). In 1994 Thalidomide was found to have anti-angiogenic activity[6] and anti-tumor activity[7] which propelled the initiation of clinical trials for cancer including multiple myeloma. The discovery of the anti-inflammatory, anti-angiogenic and anti-tumor activities of thalidomide increased the interest of further research and synthesis of safer analogs.[8][9]
Lenalidomide is the first analog of thalidomide which is marketed. It is considerably more potent than its parent drug with only two differences at a molecular level, with an added amino group at position 4 of the phthaloyl ring and removal of a carbonyl group from the phthaloyl ring.[10] Development of lenalidomide began in the late 1990s and clinical trials of lenalidomide began in 2000. In October 2001 lenalidomide was granted orphan status for the treatment of MM. In mid-2002 it entered phase II and by early 2003 phase III. In February 2003 FDA granted fast-track status to lenalidomide for the treatment of relapsed or refractory MM.[8] In 2006 it was approved for the treatment of MM along with dexamethasone and in 2007 by European Medicines Agency (EMA). In 2008, phase II trial observed efficacy in treating Non-Hodgkin's lymphoma.[11]
Pomalidomide (3-aminothalidomide) was the second thalidomide analog to enter the clinic being more potent than both of its predecessors.[12] First reported in 2001, pomalidomide was noted to directly inhibit myeloma cell proliferation and thus inhibiting MM both on the tumor and vascular compartments.[13] This dual activity of pomalidomide makes it more efficacious than thalidomide both in vitro and in vivo.[14] This effect is not related to TNF-α inhibition since potent TNF-α inhibitors such as rolipram and pentoxifylline did not inhibit myeloma cell growth nor angiogenesis.[9] Upregulation of interferon gamma, IL-2 and IL-10 have been reported for pomalidomide and may contribute to its anti-angiogenic and anti-myeloma activities.
Development
The thalidomide molecule is a synthetic derivative of glutamic acid and consists of a glutarimide ring and a phthaloyl ring (Figure 5).[15][16] Its IUPAC name is 2-(2,6-dioxopiperidin-3-yl)isoindole-1,3-dione and it has one chiral center[15] After thalidomide's selective inhibition of TNF-α had been reported, a renewed effort was put in thalidomide's clinical development. The clinical development led to the discovery of new analogs which strived to have improved activities and decreased side effects.[8][17]
Clinically, thalidomide has always been used as a racemate. Generally the S-isomer is associated with the infamous teratogenic effects of thalidomide and the R-isomer is devoid of the teratogenic properties but conveys the sedative effects,[8] however this view is highly debated and it has been argued that the animal model that these different R- and S-effects were seen in was not sensitive to the thalidomide teratogenic effects. Later reports in rabbits, which is a sensitive species, unveiled teratogenic effects from both isomers.[8][15][16][17] Moreover, thalidomide enantiomers have been shown to be interconversed in vivo due to the acidic chiral hydrogen in the asymmetric center (shown, for the EM-12 analog, in Figure 3),[16][17] so the plan to administer a purified single enantiomer to avoid the teratogenic effects will most likely be in vain.[8][15][16]
Development of lenalidomide and pomalidomide

One of the analogs of interest was made by isoindolinone replacement of the phthaloyl ring. It was given the name EM-12 (Figure 3). This replacement was thought to increase the bioavailability of the substance because of increased stability. The molecule had been reported to be an even more potent teratogenic agent than thalidomide in rats, rabbits and monkeys. Additionally, these analogs are more potent inhibitors of angiogenesis than thalidomide.[13] As well, the amino-thalidomide and amino-EM-12 were potent inhibitors of TNF-α.[16] These two analogs later got the name lenalidomide, which is the EM-12 amino analog, and pomalidomide, the thalidomide amino analog.[8]
Medical use
The primary use of IMiDs in medicine is in the treatment of cancers and autoimmune diseases (including one that is a response to the infection leprosy).[18] Indications for these agents that have received regulatory approval include:[19]
- Myelodysplastic syndrome, a precursor condition to acute myeloid leukaemia
- Erythema nodosum, a complication of leprosy
- Multiple myeloma
Off-label indications for which they seem promising treatments include:[20]
- Hodgkin's lymphoma
- Light chain-associated (AL) amyloidosis
- Primary myelofibrosis (PMF)
- Acute myeloid leukaemia (AML)
- Prostate cancer
- Metastatic renal cell carcinoma (mRCC)
Thalidomide
Thalidomide has been approved by the FDA for ENL and MM in combination with dexamethasone. EMA has also approved it to treat MM in combination with prednisone and/or melphalan. Orphan indications by the FDA include graft-versus-host disease, mycobacterial infection, recurrent aphthous ulcers, severe recurrent aphthous stomatitis, primary brain malignancies, AIDS-associated wasting syndrome, Crohn's disease, Kaposi's sarcoma, myelodysplastic syndrome and hematopoietic stem cell transplantation.[21][22]
Lenalidomide
Lenalidomide is approved in nearly 70 countries, in combination with dexamethasone for the treatment of patients with MM who have received at least one prior therapy. Orphan indications include diffuse large B-cell lymphoma, chronic lymphocytic leukemia and mantle cell lymphoma. Lenalidomide is also approved for transfusion-dependent anemia due to low or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalities in the U.S., Canada, Switzerland, Australia, New Zealand, Malaysia, Israel and several Latin American countries, while marketing authorization application is currently being evaluated in a number of other countries.[23][24] Numerous clinical trials are already in the pipeline or being conducted to explore further use for lenalidomide, alone or in combination with other drugs. Some of these indications include acute myeloid leukemia, follicular lymphoma, MALT lymphoma, Waldenström macroglobulinemia, lupus erythematosus, Hodgkin's lymphoma, myelodysplastic syndrome and more.[25][26]
Pomalidomide
Pomalidomide was submitted for FDA approval on April 26, 2012[27] and on 21 June it was announced that the drug would get standard FDA review. A marketing authorization application was filed to EMA 21 June 2012, where a decision could come as soon as early 2013. EMA has already granted pomalidomide an orphan designation for primary myelofibrosis, MM, systemic sclerosis, post-polycythaemia and post-essential thrombocythaemia myelofibrosis.[28]
Adverse effects
The major toxicities of approved IMiDs are peripheral neuropathy, thrombocytopenia, anaemia and venous thromboembolism.[20] There may be an increased risk of secondary malignancies, especially acute myeloid leukaemia in those receiving IMiDs.[20]
Teratogenicity
Thalidomide's teratogenicity has been a subject of much debate and over the years numerous hypotheses have been proposed. Two of the best-known have been the anti-angiogenesis hypothesis and oxidative stress model hypothesis, with considerable experimental evidence supporting these two hypotheses regarding thalidomide's teratogenicity.[29]
Recently, new findings have emerged that suggest a novel mechanism of teratogenicity. Cereblon is a 51 kDa protein localized in the cytoplasm, nucleus and peripheral membrane of cells in numerous parts of the body.[30] It acts as a component of the E3 ubiquitin ligase, regulating various developmental processes, including embryogenesis, carcinogenesis and cell cycle regulation, through degradation (ubiquitination) of unknown substrates. Thalidomide has been shown to bind to cereblon, inhibiting the activity of the E3 ubiquitin ligase, resulting in accumulation of the ligase substrates and downregulation of fibroblast growth factor 8 (FGF8) and FGF10. This disrupts the positive feedback loop between the two growth factors, possibly causing both multiple birth defects and anti-myeloma effects.
Findings also support the hypothesis that an increase in the expression of cereblon is an essential element of the anti-myeloma effect of both lenalidomide and pomalidomide.[29] Cereblon expression was three times higher in responding patients compared to non-responders and higher cereblon expression was also associated with partial or full response while lower expression was associated with stable or progressive disease.[30]
Mechanism of action
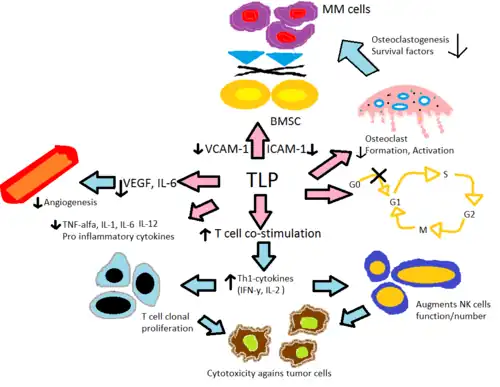
Their mechanism of action is not entirely clear, but it is known that they inhibit the production of tumour necrosis factor, interleukin 6 and immunoglobulin G and VEGF (which leads to its anti-angiogenic effects), co-stimulates T cells and NK cells and increases interferon gamma and interleukin 2 production.[31][32][33] Their teratogenic effects appear to be mediated by binding to cereblon.[34]
Thalidomide and its analogs, lenalidomide and pomalidomide, are believed to act in a similar fashion even though their exact mechanism of action is not yet fully understood. It is believed that they work through different mechanisms in various diseases. The net effect is probably due to different mechanisms combined. Mechanism of action will be explained in light of today's knowledge.
Thalidomide, lenalidomide and pomalidomide
Altering cytokine production
Thalidomide and its immune-modulating analogs alter the production of the inflammatory cytokines TNF-α, IL-1, IL-6, IL-12 and anti-inflammatory cytokine IL-10.[30] The analogs are believed to inhibit the production of TNF-α, where the analogs are up to 50.000 times more potent in vitro than the parent drug thalidomide.[35] The mechanism is believed to be through enhanced degradation of TNF-α mRNA, resulting in diminished amounts of this pro-inflammatory cytokine secreted.[36] This explains the effect of thalidomide when given to ENL patients, as they commonly have high levels of TNF-α in their blood and in dermatological lesions.[8] In contrast, in vitro assay demonstrated that TNF-α is actually enhanced in T-cell activation, where CD4+ and CD8+ T lymphocytes were stimulated by anti-CD3[8][35] which was later confirmed in an early phase trials involving solid tumors and inflammatory dermatologic diseases.[36] IL-12 is another cytokine both suppressed and enhanced by thalidomide and its analogs. When monocytes are stimulated by lipopolysaccharides, IL-12 production is suppressed but during T-cell stimulation the production is enhanced.[35]
Lenalidomide is believed to be about 1000 times more potent in vitro than thalidomide in anti-inflammatory properties and pomalidomide about 10 times more potent than lenalidomide. It is worth noticing however that, when comparing lenalidomide and pomalidomide, clinical relevance of higher in vitro potency is unclear since maximum tolerated dose of pomalidomide is 2 mg daily compared to 25 mg for lenalidomide, leading to 10-100 times lower plasma drug concentration of pomalidomide.[37]
T-cell activation
Thalidomide and its analogs help with the co-stimulation of T-cells through the B7-CD28 complex by phosphorylating tyrosine on the CD28 receptor.[8] In vitro data suggests this co-stimulation leads to increased Th1 type cytokine release of IFN-γ and IL-2 that further stimulates clonal T cell proliferation and natural killer cell proliferation and activity. This enhances natural and antibody dependent cellular cytotoxicity.[38] Lenalidomide and pomalidomide are about 100-1000 times more potent in stimulating T-cell clonal proliferation than thalidomide. In addition, in vitro data suggests pomalidomide reverts Th2 cells into Th1 by enhancing transcription factor T-bet.[30]
Anti-angiogenesis
Angiogenesis or the growth of new blood vessels has been reported to correspond with MM progression where vascular endothelial growth factor (VEGF) and its receptor, bFGF[8] and IL-6[35] appear to be required for endothelial cell migration during angiogenesis. Thalidomide and its analogs are believed to suppress angiogenesis through modulation of the above-mentioned factors where potency in anti-angiogenic activity for lenalidomide and pomalidomide was 2-3 times higher than for thalidomide in various in vivo assays,[39] Thalidomide has also been shown to block NF-κB activity through the blocking of IL-6, and NF-κB has been shown to be involved in angiogenesis.[35] Inhibition of TNF-α is not the mechanism of thalidomide's inhibition of angiogenesis since numerous other TNF-α inhibitors do not inhibit angiogenesis.[6]
Anti-tumor activity
In vivo anti-tumor activity of thalidomide is believed to be due to the potent anti-angiogenic effect and also through changes in cytokine expression. In vitro assays on apoptosis in MM cells have been shown, when treated with thalidomide and its analogs, to upregulate the activity of caspase-8. This causes cross talking of apoptotic signaling between caspase-8 and caspase-9 leading to indirect upregulation of caspase-9 activity.[30][36] Further anti-tumor activity is mediated through the inhibition of apoptosis protein-2[39] and pro-survival effects of IGF-1, increasing sensitivity to FAS mediated cell death and enhancement of TNF-related apoptosis inducing ligand.[36] They have also been shown to cause dose dependent G0/G1 cell cycle arrest in leukemia cell lines[35] where the analogs showed 100 times more potency than thalidomide.[37]
Bone marrow environment
The role of angiogenesis in the support of myeloma was first discovered by Vacca in 1994.[40] They discovered increased bone marrow angiogenesis correlates with myeloma growth and supporting stromal cells are a significant source for angiogenic molecules in myeloma. This is believed to be a main component of the mechanism in vivo by which thalidomide inhibits multiple myeloma.
Additionally, inflammatory responses within the bone marrow are believed to foster many hematological diseases. The secretion of IL-6 by bone marrow stromal cells (BMSC) and the secretion of the adhesion molecules VCAM-1, ICAM-1 and LFA, is induced in the presence of TNF-α and the adhesion of MM cells to BMSC. In vitro proliferation of MM cell lines and inhibition of Fas-mediated apoptosis is promoted by IL-6.[36] Thalidomide and its analogs directly decrease the up-regulation of IL-6 and indirectly through TNF-α, thereby reducing the secretion of adhesion molecules leading to fewer MM cells adhering to BMSC. Osteoclasts become highly active during MM, leading to bone resorption and secretion of various MM survival factors. They decrease the levels of adhesion molecules paramount to osteoclast activation, decrease the formation of the cells that form osteoclasts and downregulate cathepsin K, an important cysteine protease expressed in osteoclasts.[39]
Structure-activity relationship
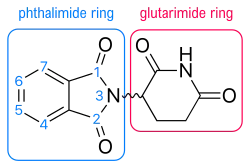
Since the mechanism of action of thalidomide and its analogs is not fully clear and the bioreceptor for these substances has not been identified, the insight into the relationship between the structure and activity of thalidomide and its analogs are mostly derived from molecular modelling and continued research investigation.[17][41] The information on SAR of thalidomide and its analogs is still in process so any trends detailed here are observed during individual studies. Research has mainly focused on improving the TNF-α and PDE4 inhibition of thalidomide,[8][15] as well as the anti-angiogenesis activity.[42][43]
TNF-α inhibitors (not via PDE4)
Research indicated that a substitution at the phthaloyl ring would increase TNF-α inhibition activity (Figure 5). An amino group substitution was tested at various locations on the phthaloyl ring (C4, C5, C6, C7) of thalidomide and EM-12 (previously described). Amino addition at the C4 location on both thalidomide and EM-12 resulted in much more potent inhibition of TNF-α. This also revealed that the amino group needed to be directly opposite the carbonyl group on the isoindolinone ring system for the most potent activity.[44] These analogs do not inhibit PDE4 and therefore do not act by PDE4 inhibition. Other additions of longer and bigger groups at the C4 and C5 position of the phthaloyl ring system of thalidomide, some with an olefin functionality, have been tested with various results. Increased inhibitory effect, compared to thalidomide, was noticed with the groups that had an oxygen atom attached directly to the C5 or C4 olefin. Iodine and bromine addition at C4 or C5 resulted in equal or decreased activity compared to thalidomide.[45] These groups were not compared with lenalidomide or pomalidomide.
PDE4 inhibitors

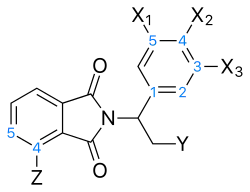
The common structure for analogs that inhibit TNF-α via inhibition of PDE4 is prepared on the basis of hydrolysing the glutarimide ring of thalidomide. These analogs do not have an acidic chiral hydrogen, unlike thalidomide, and would therefore be expected to be chirally stable.[16]
On the phenyl ring, a 3,4-dialkoxyphenyl moiety (Figure 6) is a known pharmacophore in PDE4 inhibitors such as rolipram. Optimal activity is achieved with a methoxy group at the 4-position (X2) and a bigger group, such as cyclopentoxy at the 3-position carbon (X3). However the thalidomide PDE4 inhibitory analogs do not follow the SAR of rolipram analogs directly. For thalidomide analogs, an ethoxy group at X3 and a methoxy group at X2, with X1 being just a hydrogen, gave the highest PDE4 and TNF-α inhibition.[15] Substitutes larger than diethoxy at the X2–X3 position had decreased activity. The effects of these substitutions seem to be mediated by steric effects.[16]
For the Y-position, a number of groups have been explored. Substituted amides that were larger than methylamide (CONHCH3) decrease PDE4 inhibition activity.[16] Using a carboxylic acid as a starting point, an amide group has similar PDE4 inhibition activity but both groups were shown to be a considerably less potent than a methyl ester group, which had about six-fold increase in PDE4 inhibitory activity. Sulfone group had similar PDE4 inhibition as the methyl ester group. The best PDE4 inhibition was observed when a nitrile group was attached, which has 32 times more PDE4 inhibitory activity than the carboxyl acid.[15] Substituents at Y leading to increasing PDE4 inhibitory activity thus followed the order:
- COOH ≤ CONH2 ≤ COOCH3 ≤ SO2CH3 < CN
Substitutions on the phthaloyl ring have been explored and it was noticed that nitro groups at the C4 or C5 location decreased activity but C4 or C5 amino substitution increased it dramatically.[16] When the substitution at the 4 (Z) location on the phthaloyl ring was examined, hydroxyl and methoxy groups seem to make the analog a less potent PDE4 inhibitor. An increase in activity was observed with amino and dimethylamino to a similar extent but a methyl group improved the activity further than the aforementioned groups. A 4-N-acetylamino group had slightly lower PDE4 inhibitory activity, compared with the methyl group, but increased the compound's TNF-α inhibitory activity to a further extent.[15] Substituents at Z leading to increasing PDE4 inhibitory activity thus followed the order:
- N(CH3)2 ≤ NH2 < NHC(O)CH3 < CH3
Angiogenesis inhibition
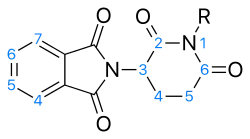
For angiogenesis inhibition activity, an intact glutarimide ring seems to be required. Different groups were tested in the R position. The substances that had nitrogen salts as the R group showed good activity. The improved angiogenesis inhibitory activity could be due to increased solubility or that the positively charged nitrogen has added interaction with the active site. Tetrafluorination of the phthaloyl ring seems to increase the angiogenesis inhibition.[42]
Synthesis
Described below are schemes for synthesizing thalidomide, lenalidomide, and pomalidomide, as reported from prominent primary literature. Note that these synthesis schemes do not necessarily reflect the organic synthesis strategies used to synthesize these single chemical entities.
Thalidomide


Synthesis of thalidomide has usually been performed as seen in scheme 1. This synthesis is a reasonably simplistic three step process. The downside of this process however is that the last step requires a high-temperature melt reaction which demands multiple recrystallizations and is not compliant with standard equipment.
Scheme 2 is the newer synthesis route which was designed to make the reaction more direct and to produce better yields. This route uses L-glutamine rather than L-glutamic acid as a starting material and by letting it react with N-carbethoxyphthalimide gives N-phthaloyl-L-glutamine (4), with 50–70% yield. The substance 4 is then stirred in a mixture with carbonyldiimidazole (CDI) with enough 4-dimethylaminopyridine (DMAP) in tetrahydrofuran (THF) to catalyze the reaction and heated to reflux for 15–18 hours. During the reflux thalidomide crystallizes out of the mixture. The final step gives 85–93% yield of thalidomide, bringing the total yield to 43–63%.[46]
Lenalidomide and pomalidomide

Both of the amino analogs are prepared from the condensation of 3-aminopiperidine-2,6-dione hydrochloride (Compound 3) which is synthesized in a two step reaction from commercially available Cbz-L-glutamine. The Cbz-L-glutamine is treated with CDI in refluxing THF to yield Cbz-aminoglutarimide. To remove the Cbz protecting group hydrogenolysis, under 50–60 psi of hydrogen with 10% Pd/C mixed with ethyl acetate and HCl, was performed. The formulated hydrochloride (Compound 3 in Scheme 3) was then reacted with 3-nitrophthalic anhydride in refluxing acetic acid to yield the 4-nitro substituted thalidomide analog and the nitro group then reduced with hydrogenation to give pomalidomide.[44]
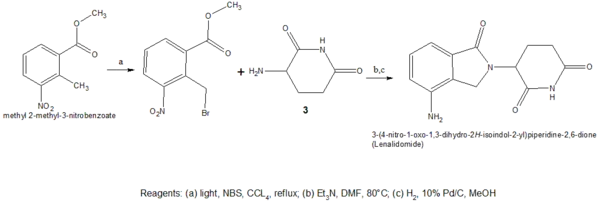
Lenalidomide is synthesized in a similar way using compound 3 (3-aminopiperidine-2,6-dione) treated with a nitro-substituted methyl 2-(bromomethyl) benzoate, and hydrogenation of the nitro group.[44]
Pharmacokinetics
Thalidomide
| Thalidomide | ||
|---|---|---|
| Tmax [drug] | 4–6 hours in subjects with MM[47] |
|
| Protein binding | 55–65%[48] | |
| Metabolites | Hydrolized metabolites[48] | |
| Half-life [t1/2] | 5.5–7.6 hours[48] | |
Lenalidomide
| Lenalidomide | ||
|---|---|---|
| Tmax [drug] | 0.6–1.5 hours in healthy subjects[49] 0.5–4 hours in subjects with MM[50] |
 |
| Protein binding | ~30%[49] | |
| Metabolites | Has not yet been studied[49] | |
| Half-life [t1/2] | 3 hours in healthy subjects[49] 3.1–4.2 hours in subjects with MM[50] | |
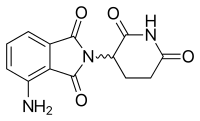
Pomalidomide
| Pomalidomide | ||
|---|---|---|
| Tmax [drug] | 0.5–8 hours[51] |
 |
| Protein binding | Unknown | |
| Metabolites | Unknown | |
| Half-life [t1/2] | 6.2–7.9 hours[51] | |
See also
- Cancer
- Multiple myeloma
- Drug design
- Thalidomide
- Lenalidomide
- Pomalidomide
- Apremilast
- Organic chemistry
- Health crisis
- Immunomodulation therapy
- Immunosuppressant
- Immunomodulatory drug
References
- 1 2 Knight, R (August 2005). "IMiDs: a novel class of immunomodulators". Seminars in Oncology. 32 (4 Suppl 5): S24–S30. doi:10.1053/j.seminoncol.2005.06.018. PMID 16085014.
- ↑ Aragon-Ching AB, Li H, Gardner ER, Figg WD (2007). "Thalidomide analogues as anticancer drugs". Recent Pat Anti-Cancer Drug Discov. 2 (2): 167–174. doi:10.2174/157489207780832478. PMC 2048745. PMID 17975653.
- ↑ Reversal of Fortune: How a Vilified Drug Became a Life-saving Agent in the "War" Against Cancer - Onco'Zine - The International Oncology Network (November 30, 2013) Archived January 3, 2014, at archive.today
- ↑ Mazzoccoli, L; Cadoso, SH; Amarante, GW; de Souza, MV; Domingues, R; Machado, MA; de Almeida, MV; Teixeira, HC (July 2012). "Novel thalidomide analogues from diamines inhibit pro-inflammatory cytokine production and CD80 expression while enhancing IL-10". Biomedicine & Pharmacotherapy. 66 (5): 323–9. doi:10.1016/j.biopha.2012.05.001. PMID 22770990.
- ↑ Prommer, E. E. (20 October 2009). "Review Article: Palliative Oncology: Thalidomide". American Journal of Hospice and Palliative Medicine. 27 (3): 198–204. doi:10.1177/1049909109348981. PMID 19843880. S2CID 24167431.
- 1 2 D'Amato RJ, Loughnan MS, Flynn E, Folkman J (April 1994). "Thalidomide is an inhibitor of angiogenesis". Proc. Natl. Acad. Sci. U.S.A. 91 (9): 4082–5. Bibcode:1994PNAS...91.4082D. doi:10.1073/pnas.91.9.4082. PMC 43727. PMID 7513432.
- ↑ Verheul HM, Panigrahy D, Yuan J, D'Amato RJ (January 1999). "Combination oral antiangiogenic therapy with thalidomide and sulindac inhibits tumour growth in rabbits". Br. J. Cancer. 79 (1): 114–8. doi:10.1038/sj.bjc.6690020. PMC 2362163. PMID 10408702.
- 1 2 3 4 5 6 7 8 9 10 11 12 Bartlett, J. Blake; Dredge, Keith; Dalgleish, Angus G. (1 April 2004). "Timeline: The evolution of thalidomide and its IMiD derivatives as anticancer agents". Nature Reviews Cancer. 4 (4): 314–322. doi:10.1038/nrc1323. PMID 15057291. S2CID 7293027.
- 1 2 D'Amato RJ, Lentzsch S, Anderson KC, Rogers MS (December 2001). "Mechanism of action of thalidomide and 3-aminothalidomide in multiple myeloma". Semin. Oncol. 28 (6): 597–601. doi:10.1016/S0093-7754(01)90031-4. PMID 11740816.
- ↑ Zimmerman, Todd (1 May 2009). "Immunomodulatory agents in oncology". Update on Cancer Therapeutics. 3 (4): 170–181. doi:10.1016/j.uct.2009.03.003.
- ↑ Zeldis, Jerome B.; Knight, Robert; Hussein, Mohamad; Chopra, Rajesh; Muller, George (1 March 2011). "A review of the history, properties, and use of the immunomodulatory compound lenalidomide". Annals of the New York Academy of Sciences. 1222 (1): 76–82. Bibcode:2011NYASA1222...76Z. doi:10.1111/j.1749-6632.2011.05974.x. PMID 21434945. S2CID 5336195.
- ↑ "Vector has moved".
- 1 2 D'Amato, RJ; Lentzsch, S; Anderson, KC; Rogers, MS (December 2001). "Mechanism of action of thalidomide and 3-aminothalidomide in multiple myeloma". Seminars in Oncology. 28 (6): 597–601. doi:10.1016/S0093-7754(01)90031-4. PMID 11740816.
- ↑ Lentzsch S, Rogers MS, LeBlanc R, et al. (April 2002). "S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice". Cancer Res. 62 (8): 2300–5. PMID 11956087.
- 1 2 3 4 5 6 7 8 Man, Hon-Wah; Schafer, Peter; Wong, Lu Min; Patterson, Rebecca T.; Corral, Laura G.; Raymon, Heather; Blease, Kate; Leisten, Jim; Shirley, Michael A.; Tang, Yang; Babusis, Darius M.; Chen, Roger; Stirling, Dave; Muller, George W. (26 March 2009). "Discovery of (S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide (Apremilast), a Potent and Orally Active Phosphodiesterase 4 and Tumor Necrosis Factor-α Inhibitor". Journal of Medicinal Chemistry. 52 (6): 1522–4. doi:10.1021/jm900210d. PMID 19256507.
- 1 2 3 4 5 6 7 8 9 Muller, George W.; Corral, Laura G.; Shire, Mary G.; Wang, Hua; Moreira, Andre; Kaplan, Gilla; Stirling, David I. (1 January 1996). "Structural Modifications of Thalidomide Produce Analogs with Enhanced Tumor Necrosis Factor Inhibitory Activity". Journal of Medicinal Chemistry. 39 (17): 3238–3240. doi:10.1021/jm9603328. PMID 8765505.
- 1 2 3 4 Man, Hon-Wah; Corral, Laura G; Stirling, David I; Muller, George W (1 October 2003). "α-Fluoro-substituted thalidomide analogues". Bioorganic & Medicinal Chemistry Letters. 13 (20): 3415–3417. doi:10.1016/S0960-894X(03)00778-9. PMID 14505639.
- ↑ Pan, B; Lentzsch, S (October 2012). "The application and biology of immunomodulatory drugs (IMiDs) in cancer". Pharmacology & Therapeutics. 136 (1): 56–68. doi:10.1016/j.pharmthera.2012.07.004. PMID 22796518.
- ↑ Sedlarikova, L; Kubiczkova, L; Sevcikova, S; Hajek, R (October 2012). "Mechanism of immunomodulatory drugs in multiple myeloma". Leukemia Research. 36 (10): 1218–1224. doi:10.1016/j.leukres.2012.05.010. PMID 22727252.
- 1 2 3 Vallet, S; Witzens-Harig, M; Jaeger, D; Podar, K (March 2012). "Update on immunomodulatory drugs (IMiDs) in hematologic and solid malignancies". Expert Opinion on Pharmacotherapy. 13 (4): 473–494. doi:10.1517/14656566.2012.656091. PMID 22324734. S2CID 7981368.
- ↑ "Thalomid (Thalidomide) dosing, indications, interactions, adverse effects, and more". MedScape reference. Retrieved 18 September 2012.
- ↑ "Thalidomide Celgene (previously Thalidomide Pharmion)". European Medicines Agency. Retrieved 18 September 2012.
- ↑ "Celgene Biopharmaceutical - Investor relations - Press Releases". Archived from the original on 19 January 2013. Retrieved 18 September 2012.
- ↑ "Revlimid (lenalidomide) dosing, indications, interactions, adverse effects, and more". Medscape references. Retrieved 18 September 2012.
- ↑ "Search of: lenalidomide - List Results". Clinical Trials. Retrieved 18 September 2012.
- ↑ "Clinical Trials Register". EU Clinical Trials Register. Retrieved 18 September 2012.
- ↑ "Celgene Submits Pomalidomide For FDA Approval". The myeloma beacon.
- ↑ "European Medicines Agency - Search results from your query". European Medicines Agency. Retrieved 18 September 2012.
- 1 2 Ito, Takumi; Handa, Hiroshi (1 March 2012). "Deciphering the mystery of thalidomide teratogenicity". Congenital Anomalies. 52 (1): 1–7. doi:10.1111/j.1741-4520.2011.00351.x. PMID 22348778.
- 1 2 3 4 5 Martiniani, Roberta; Di Loreto, Valentina; Di Sano, Chiara; Lombardo, Alessandra; Liberati, Anna Marina (1 January 2012). "Biological Activity of Lenalidomide and Its Underlying Therapeutic Effects in Multiple Myeloma". Advances in Hematology. 2012: 842945. doi:10.1155/2012/842945. PMC 3417169. PMID 22919394.
- ↑ Quach, H; Ritchie, D; Stewart, AK; Neeson, P; Harrison, S; Smyth, MJ; Prince, HM (January 2010). "Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma". Leukemia. 24 (1): 22–32. doi:10.1038/leu.2009.236. PMC 3922408. PMID 19907437.
- ↑ Andhavarapu, S; Roy, V (February 2013). "Immunomodulatory drugs in multiple myeloma". Expert Review of Hematology. 6 (1): 69–82. doi:10.1586/ehm.12.62. PMID 23373782. S2CID 12782141.
- ↑ Sedlarikova, L; Kubiczkova, L; Sevcikova, S; Hajek, R (October 2012). "Mechanism of immunomodulatory drugs in multiple myeloma". Leukemia Research. 36 (10): 1218–1224. doi:10.1016/j.leukres.2012.05.010. PMID 22727252.
- ↑ Chang, XB; Stewart, AK (2011). "What is the functional role of the thalidomide binding protein cereblon?". International Journal of Biochemistry and Molecular Biology. 2 (3): 287–94. PMC 3193296. PMID 22003441.
- 1 2 3 4 5 6 Huang, Yen-Ta; Hsu, Chih W.; Chiu, Ted H. (1 September 2008). "Thalidomide and Its Analogs as Anticancer Agents". Tzu Chi Medical Journal. 20 (3): 188–195. doi:10.1016/S1016-3190(08)60034-8.
- 1 2 3 4 5 Melchert, Magda; List, Alan (1 July 2007). "The thalidomide saga". The International Journal of Biochemistry & Cell Biology. 39 (7–8): 1489–1499. doi:10.1016/j.biocel.2007.01.022. PMID 17369076.
- 1 2 Quach, H; Ritchie, D; Stewart, A K; Neeson, P; Harrison, S; Smyth, M J; Prince, H M (12 November 2009). "Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma". Leukemia. 24 (1): 22–32. doi:10.1038/leu.2009.236. PMC 3922408. PMID 19907437.
- ↑ Thomas, Sheeba K.; Richards, Tiffany A.; Weber, Donna M. (1 December 2007). "Lenalidomide in multiple myeloma". Best Practice & Research Clinical Haematology. 20 (4): 717–735. doi:10.1016/j.beha.2007.09.002. PMID 18070715.
- 1 2 3 Kotla, Venumadhav; Goel, Swati; Nischal, Sangeeta; Heuck, Christoph; Vivek, Kumar; Das, Bhaskar; Verma, Amit (1 January 2009). "Mechanism of action of lenalidomide in hematological malignancies". Journal of Hematology & Oncology. 2 (1): 36. doi:10.1186/1756-8722-2-36. PMC 2736171. PMID 19674465.
- ↑ Vacca A, Ribatti D, Roncali L, et al. (July 1994). "Bone marrow angiogenesis and progression in multiple myeloma". Br. J. Haematol. 87 (3): 503–8. doi:10.1111/j.1365-2141.1994.tb08304.x. PMC 3301416. PMID 7527645.
- ↑ Avila, Carolina Martins; Romeiro, Nelilma Correia; Sperandio da Silva, Gilberto M.; Sant’Anna, Carlos M.R.; Barreiro, Eliezer J.; Fraga, Carlos A.M. (1 October 2006). "Development of new CoMFA and CoMSIA 3D-QSAR models for anti-inflammatory phthalimide-containing TNFα modulators". Bioorganic & Medicinal Chemistry. 14 (20): 6874–6885. doi:10.1016/j.bmc.2006.06.042. PMID 16843662.
- 1 2 Lepper, Erin R.; Ng, Sylvia S. W.; Gütschow, Michael; Weiss, Michael; Hauschildt, Sunna; Hecker, Thomas K.; Luzzio, Frederick A.; Eger, Kurt; Figg, William D. (1 April 2004). "Comparative Molecular Field Analysis and Comparative Molecular Similarity Indices Analysis of Thalidomide Analogues as Angiogenesis Inhibitors". Journal of Medicinal Chemistry. 47 (9): 2219–2227. doi:10.1021/jm0304820. PMID 15084120.
- ↑ Noguchi, Tomomi; Fujimoto, Haruka; Sano, Hiroko; Miyajima, Atsushi; Miyachi, Hiroyuki; Hashimoto, Yuichi (1 December 2005). "Angiogenesis inhibitors derived from thalidomide". Bioorganic & Medicinal Chemistry Letters. 15 (24): 5509–5513. doi:10.1016/j.bmcl.2005.08.086. PMID 16183272.
- 1 2 3 Muller, GW; Chen, R; Huang, SY; Corral, LG; Wong, LM; Patterson, RT; Chen, Y; Kaplan, G; Stirling, DI (Jun 7, 1999). "Amino-substituted thalidomide analogs: potent inhibitors of TNF-alpha production". Bioorganic & Medicinal Chemistry Letters. 9 (11): 1625–30. doi:10.1016/s0960-894x(99)00250-4. PMID 10386948.
- ↑ Stewart, Scott G.; Spagnolo, Daniel; Polomska, Marta E.; Sin, Melvin; Karimi, Mahdad; Abraham, Lawrence J. (1 November 2007). "Synthesis and TNF expression inhibitory properties of new thalidomide analogues derived via Heck cross coupling". Bioorganic & Medicinal Chemistry Letters. 17 (21): 5819–5824. doi:10.1016/j.bmcl.2007.08.042. PMID 17851074.
- ↑ Muller, George W.; Konnecke, William E.; Smith, Alison M.; Khetani, Vikram D. (1 March 1999). "A Concise Two-Step Synthesis of Thalidomide". Organic Process Research & Development. 3 (2): 139–140. doi:10.1021/op980201b.
- ↑ Chung, F. (1 September 2004). "Thalidomide Pharmacokinetics and Metabolite Formation in Mice, Rabbits, and Multiple Myeloma Patients". Clinical Cancer Research. 10 (17): 5949–5956. doi:10.1158/1078-0432.CCR-04-0421. PMID 15355928.
- 1 2 3 "Summary of product characteristics: Thalidomid Celgene" (PDF). European Medicines Agency. Retrieved 23 September 2012.
- 1 2 3 4 Armoiry, X.; Aulagner, G.; Facon, T. (1 June 2008). "Lenalidomide in the treatment of multiple myeloma: a review". Journal of Clinical Pharmacy and Therapeutics. 33 (3): 219–226. doi:10.1111/j.1365-2710.2008.00920.x. PMID 18452408.
- 1 2 Richardson, P. G. (12 July 2002). "Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma". Blood. 100 (9): 3063–3067. doi:10.1182/blood-2002-03-0996. PMID 12384400.
- 1 2 Schey, S.A. (15 August 2004). "Phase I Study of an Immunomodulatory Thalidomide Analog, CC-4047, in Relapsed or Refractory Multiple Myeloma". Journal of Clinical Oncology. 22 (16): 3269–3276. doi:10.1200/JCO.2004.10.052. PMID 15249589.