Duchenne muscular dystrophy
Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy that primarily affects boys.[3] Muscle weakness usually begins around the age of four, and worsens quickly.[2] Muscle loss typically occurs first in the thighs and pelvis followed by the arms.[3] This can result in trouble standing up.[3] Most are unable to walk by the age of 12.[2] Affected muscles may look larger due to increased fat content.[3] Scoliosis is also common.[3] Some may have intellectual disability.[3] Females with a single copy of the defective gene may show mild symptoms.[3]
| Duchenne muscular dystrophy | |
|---|---|
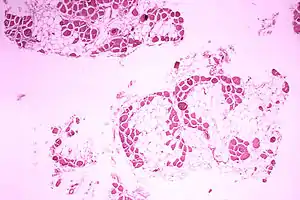 | |
| Microscopic image of cross-sectional calf muscle from a person with Duchenne muscular dystrophy, showing extensive replacement of muscle fibers by fat cells. | |
| Pronunciation |
|
| Specialty | Pediatric neurology, neuromuscular medicine, medical genetics |
| Symptoms | Muscle weakness, trouble standing up, scoliosis[2][3] |
| Usual onset | Around age 4[2] |
| Causes | Genetic (X-linked recessive)[3] |
| Diagnostic method | Genetic testing[3] |
| Treatment | Pharmacological treatment, physical therapy, braces, speech therapy, occupational therapy, surgery, assisted ventilation[2][3] |
| Medication | Corticosteroids |
| Prognosis | life expectancy: 26 years[4] |
| Frequency | In males, 1 in 3,500-6,000[3] In females, 1 in 50,000,000[5] |
The disorder is X-linked recessive.[3] About two thirds of cases are inherited from a person's mother, while one third of cases are due to a new mutation.[3] It is caused by a mutation in the gene for the protein dystrophin.[3] Dystrophin is important to maintain the muscle fiber's cell membrane.[3] Genetic testing can often make the diagnosis at birth.[3] Those affected also have a high level of creatine kinase in their blood.[3]
Although there is no known cure, physical therapy, braces, and corrective surgery may help with some symptoms.[2] Assisted ventilation may be required in those with weakness of breathing muscles.[3] Medications used include steroids to slow muscle degeneration, anticonvulsants to control seizures and some muscle activity, and immunosuppressants to delay damage to dying muscle cells.[2] Gene therapy, as a treatment, is in the early stages of study in humans.[3] A small initial study using gene therapy has given some children improved muscle strength, but long term effects are unknown as of 2020.[6]
DMD affects about one in 3,500 to 6,000 males at birth.[3] It is the most common type of muscular dystrophy.[3] The life expectancy is 26;[4] however, with excellent care, some may live into their 30s or 40s.[3] The disease is much more rare in girls, occurring approximately once in 50,000,000 live female births.[5]
Signs and symptoms
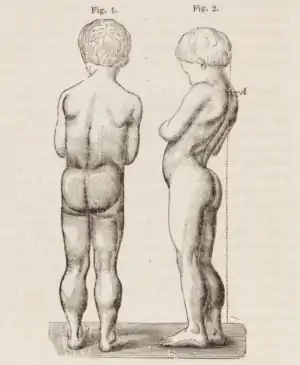
DMD causes progressive muscle weakness due to muscle fiber disarray, death, and replacement with connective tissue or fat.[3] The voluntary muscles are affected first, especially those of the hips, pelvic area, thighs, calves.[7][8] It eventually progresses to the shoulders and neck, followed by arms, respiratory muscles, and other areas.[8] Fatigue is common.[9]
Signs usually appear before age five, and may even be observed from the moment a boy takes his first steps.[10] There is general difficulty with motor skills, which can result in an awkward manner of walking, stepping, or running.[11] They tend to walk on their toes,[11] in part due to shortening of the Achilles tendon,[12] and because it compensates for knee extensor weakness.[8] Falls can be frequent.[13] It becomes harder and harder for the boy to walk; his ability to walk usually completely disintegrates before age 13.[11] Most men affected with DMD become essentially "paralyzed from the neck down" by the age of 21.[10] Cardiomyopathy, particularly dilated cardiomyopathy, is common, seen in half of 18-year-olds.[11] The development of congestive heart failure or arrhythmia (irregular heartbeat) is only occasional.[8] In late stages of the disease, respiratory impairment and swallowing impairment can occur, which can result in pneumonia.[14]

A classic sign of DMD is trouble getting up from lying or sitting position,[13] as manifested by a positive Gowers's sign. When a child tries to arise from lying on his stomach, he compensates for pelvic muscle weakness through use of the upper extremities:[11] first by rising to stand on his arms and knees, and then "walking" his hands up his legs to stand upright. Another characteristic sign of DMD is pseudohypertrophy (enlarging) of the muscles of the tongue, calves, buttocks, and shoulders (around age 4 or 5). The muscle tissue is eventually replaced by fat and connective tissue, hence the term pseudohypertrophy. Muscle fiber deformities and muscle contractures of Achilles tendon and hamstrings can occur, which impair functionality because the muscle fibers shorten and fibrose in connective tissue.[8] Skeletal deformities can occur, such as lumbar hyperlordosis, scoliosis, anterior pelvic tilt, and chest deformities. Lumbar hyperlordosis is thought to be compensatory mechanism in response to gluteal and quadriceps muscle weakness, all of which cause altered posture and gait (e.g.: restricted hip extension).[15][16]
Non musculoskeletal manifestations of DMD occur. There is a higher risk of neurobehavioral disorders (e.g., ADHD), learning disorders (dyslexia), and non-progressive weaknesses in specific cognitive skills (in particular short-term verbal memory),[11] which are believed to be the result of absent or dysfunctional dystrophin in the brain.[17]
Cause
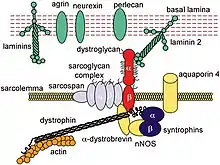
DMD is caused by a mutation of the dystrophin gene, located on the short arm of the X chromosome (locus Xp21)[18] that codes for dystrophin protein. Mutations can either be inherited or occur spontaneously during germline transmission, causing to a large reduction or absence of dystrophin, a protein that provides structural integrity in muscle cells.[19] Dystrophin is responsible for connecting the actin cytoskeleton of each muscle fiber to the underlying basal lamina (extracellular matrix), through a protein complex containing many subunits. The absence of dystrophin permits excess calcium to penetrate the sarcolemma (the cell membrane).[20] Alterations in calcium and signalling pathways cause water to enter into the mitochondria, which then burst.
In skeletal muscle dystrophy, mitochondrial dysfunction gives rise to an amplification of stress-induced cytosolic calcium signals and an amplification of stress-induced reactive-oxygen species production. In a complex cascading process that involves several pathways and is not clearly understood, increased oxidative stress within the cell damages the sarcolemma and eventually results in the death of the cell. Muscle fibers undergo necrosis and are ultimately replaced with adipose and connective tissue.
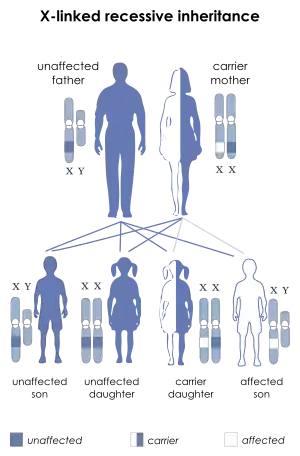
DMD is inherited in an X-linked recessive pattern. Females typically are carriers of the genetic trait while males are affected. A female carrier will be unaware she carries a mutation until she has an affected son. The son of a carrier mother has a 50% chance of inheriting the defective gene from his mother. The daughter of a carrier mother has a 50% chance of being a carrier and a 50% chance of having two normal copies of the gene. In all cases, an unaffected father either passes a normal Y to his son or a normal X to his daughter. Female carriers of an X-linked recessive condition, such as DMD, can show symptoms depending on their pattern of X-inactivation.
DMD is extremely rare in females (about 1 in 50,000,000 female births).[5] It can occur in females with an affected father and a carrier mother, in those who are missing an X chromosome, or those who have an inactivated X chromosome (the most common of the rare reasons).[21] The daughter of a carrier mother and an affected father will be affected or a carrier with equal probability, as she will always inherit the affected X-chromosome from her father and has a 50% chance of also inheriting the affected X-chromosome from her mother.[22]
Disruption of the blood-brain barrier has been seen to be a noted feature in the development of DMD.[23]
Diagnosis
Genetic counseling is advised for people with a family history of the disorder. DMD can be detected with about 95% accuracy by genetic studies performed during pregnancy.[14] Creatine kinase (CPK-MM) levels in the bloodstream are extremely high. An electromyography (EMG) shows that weakness is caused by destruction of muscle tissue rather than by damage to nerves.
DNA test
The muscle-specific isoform of the dystrophin gene is composed of 79 exons, and DNA testing (blood test) and analysis can usually identify the specific type of mutation of the exon or exons that are affected. DNA testing confirms the diagnosis in most cases.[24]
Muscle biopsy
If DNA testing fails to find the mutation, a muscle biopsy test may be performed.[25] A small sample of muscle tissue is extracted using a biopsy needle. The key tests performed on the biopsy sample for DMD are immunohistochemistry, immunocytochemistry, and immunoblotting for dystrophin, and should be interpreted by an experienced neuromuscular pathologist.[26] These tests provide information on the presence or absence of the protein. Absence of the protein is a positive test for DMD. Where dystrophin is present, the tests indicate the amount and molecular size of dystrophin, helping to distinguish DMD from milder dystrophinopathy phenotypes.[27] Over the past several years, DNA tests have been developed that detect more of the many mutations that cause the condition, and muscle biopsy is not required as often to confirm the presence of DMD.[28]
Prenatal tests
A prenatal test can be considered when the mother is a known or suspected carrier.[29]
Prenatal tests can tell whether the unborn child has one of the most common mutations. Many mutations are responsible for DMD, and some have not been identified, so genetic testing may be falsely negative if the suspected mutation in the mother has not been identified.
Prior to invasive testing, determination of the fetal sex is important; while males are sometimes affected by this X-linked disease, female DMD is extremely rare. This can be achieved by ultrasound scan at 16 weeks or more recently by free fetal DNA (cffDNA) testing. Chorion villus sampling (CVS) can be done at 11–14 weeks, and has a 1% risk of miscarriage. Amniocentesis can be done after 15 weeks, and has a 0.5% risk of miscarriage. Non invasive prenatal testing can be done around 10–12 weeks.[30] Another option in the case of unclear genetic test results is fetal muscle biopsy.
Treatment
No cure for DMD is known, and an ongoing medical need has been recognized by regulatory authorities.[31] Gene therapy has shown some success.[32]
Treatment is generally aimed at controlling symptoms to maximize the quality of life which can be measured using specific questionnaires,[33] and include:
- Corticosteroids such as prednisolone and deflazacort lead to short-term improvements in muscle strength and function up to 2 years.[34] Corticosteroids have also been reported to help prolong walking, though the evidence for this is not robust.[35]
- Randomised control trials have shown that β2 agonists increase muscle strength, but do not modify disease progression. Follow-up time for most RCTs on β2 agonists is only around 12 months, hence results cannot be extrapolated beyond that time frame.
- Mild, nonjarring physical activity such as swimming is encouraged. Inactivity (such as bed rest) can worsen the muscle disease.
- Physical therapy is helpful to maintain muscle strength, flexibility, and function.
- Orthopedic appliances (such as braces and wheelchairs) may improve mobility and the ability for self-care. Form-fitting removable leg braces that hold the ankle in place during sleep can defer the onset of contractures.
- Appropriate respiratory support as the disease progresses is important.
- Cardiac problems may require a pacemaker.[36]
The medication eteplirsen, a Morpholino antisense oligo, has been approved in the United States for the treatment of mutations amenable to dystrophin exon 51 skipping. The US approval has been controversial[37] as eteplirsen failed to establish a clinical benefit;[38] it has been refused approval by the European Medicines Agency.[39]
The medication ataluren (Translarna) is approved use in the European Union.[40][41]
The antisense oligonucleotide golodirsen (Vyondys 53) was approved for medical use in the United States in 2019, for the treatment of cases that can benefit from skipping exon 53 of the dystrophin transcript.[42][43]
The Morpholino antisense oligonucleotide viltolarsen (Viltepso) was approved for medical use in the United States in August 2020, for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the DMD gene that is amenable to exon 53 skipping.[44] It is the second approved targeted treatment for people with this type of mutation in the United States.[44] Approximately 8% of people with DMD have a mutation that is amenable to exon 53 skipping.[44]
Casimersen was approved for medical use in the United States in February 2021,[45] and it is the first FDA-approved targeted treatment for people who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping.[45]
Comprehensive multidisciplinary care guidelines for DMD have been developed by the Centers for Disease Control and Prevention, and were published in two parts in The Lancet Neurology in 2010.[25] An update was published in 2018.[46][47]
Physical therapy
Physical therapists are concerned with enabling patients to reach their maximum physical potential. Their aim is to:[48]
- minimize the development of contractures and deformity by developing a programme of stretches and exercises where appropriate
- anticipate and minimize other secondary complications of a physical nature by recommending bracing and durable medical equipment[49]
- monitor respiratory function and advise on techniques to assist with breathing exercises and methods of clearing secretions[48]
Respiration assistance

1 – Vocal folds
2 – Thyroid cartilage
3 – Cricoid cartilage
4 – Tracheal rings
5 – Balloon cuff
Modern "volume ventilators/respirators", which deliver an adjustable volume (amount) of air to the person with each breath, are valuable in the treatment of people with muscular dystrophy-related respiratory problems. The ventilator may require an invasive endotracheal or tracheotomy tube through which air is directly delivered, but for some people, noninvasive delivery through a face mask or mouthpiece is sufficient. Positive airway pressure machines, particularly bilevel ones, are sometimes used in this latter way. The respiratory equipment may easily fit on a ventilator tray on the bottom or back of a power wheelchair with an external battery for portability.
Ventilator treatment may start in the mid- to late teens when the respiratory muscles can begin to collapse. If the vital capacity has dropped below 40% of normal, a volume ventilator/respirator may be used during sleeping hours, a time when the person is most likely to be underventilating (hypoventilating). Hypoventilation during sleep is determined by a thorough history of sleep disorder with an oximetry study and a capillary blood gas (see pulmonary function testing). A cough assist device can help with excess mucus in lungs by hyperinflation of the lungs with positive air pressure, then negative pressure to get the mucus up. If the vital capacity continues to decline to less than 30 percent of normal, a volume ventilator/respirator may also be needed during the day for more assistance. The person gradually will increase the amount of time using the ventilator/respirator during the day as needed. However, there are also people with the disease in their 20s who have no need for a ventilator.
Future developments
There is no cure for any of the muscular dystrophies.[50] Several drugs designed to address the root cause are under development, including gene therapy (Microdystrophin), and antisense drugs (Ataluren, Eteplirsen etc.).[51] Other medications used include corticosteroids (Deflazacort), calcium channel blockers (Diltiazem) to slow skeletal and cardiac muscle degeneration, anticonvulsants to control seizures and some muscle activity, and immunosuppressants (Vamorolone) to delay damage to dying muscle cells.[2] Physical therapy, braces, and corrective surgery may help with some symptoms[2] while assisted ventilation may be required in those with weakness of breathing muscles.[3] Outcomes depend on the specific type of disorder.[52][51]
A paper published by Stanford University on 10 March 2022 demonstrated that patients with muscular dystrophies could benefit from new therapies targeting the specific pathways contributing directly to muscle disorders. Three recent advances are likely to enhance the landscape of treatments for muscular dystrophies such as DMD. First, induced pluripotent stem cells (iPSCs) allow researchers to design effective treatment strategies. Second, artificial intelligence (AI) can help identify therapeutic targets. Third, a high volume of multi-omics data gathered from diverse sources through disease models can provide valuable information about converging and diverging pathways.[19]
Prognosis
Duchenne muscular dystrophy is a rare progressive disease which eventually affects all voluntary muscles and involves the heart and breathing muscles in later stages. Life expectancy is estimated to be around 25–26,[14][4] but this varies. With excellent medical care, affected men often live into their 30s.[53] David Hatch of Paris, Maine, may be the oldest person in the world with the disease; as of 2021, he was 58.[54]
The most common direct cause of death in people with DMD is respiratory failure. Complications from treatment, such as mechanical ventilation and tracheotomy procedures, are also a concern. The next leading cause of death is cardiac-related conditions such as heart failure brought on by dilated cardiomyopathy. With respiratory assistance, the median survival age can reach up to 40. In rare cases, people with DMD have been seen to survive into their forties or early fifties, with proper positioning in wheelchairs and beds, and the use of ventilator support (via tracheostomy or mouthpiece), airway clearance, and heart medications.[55] Early planning of the required supports for later-life care has shown greater longevity for people with DMD.[56]
Curiously, in the mdx mouse model of Duchenne muscular dystrophy, the lack of dystrophin is associated with increased calcium levels and skeletal muscle myonecrosis. The intrinsic laryngeal muscles (ILMs) are protected and do not undergo myonecrosis.[57] ILMs have a calcium regulation system profile suggestive of a better ability to handle calcium changes in comparison to other muscles, and this may provide a mechanistic insight for their unique pathophysiological properties.[58] The ILM may facilitate the development of novel strategies for the prevention and treatment of muscle wasting in a variety of clinical scenarios.[59] In addition, patients with Duchenne muscular dystrophy also have elevated plasma lipoprotein levels, implying a primary state of dyslipidemia in patients.[60]
Epidemiology
DMD is the most common type of muscular dystrophy; it affects about one in 5,000 males at birth.[3] DMD has an incidence of one in 3,600 male infants.[14]
In the US, a 2010 study showed a higher amount of those with DMD age ranging from 5 to 54 who are Hispanic compared to non-Hispanic Whites, and non-Hispanic Blacks.[61]
History

The disease was first described by the Neapolitan physician Giovanni Semmola in 1834 and Gaetano Conte in 1836.[62][63][64] However, DMD is named after the French neurologist Guillaume-Benjamin-Amand Duchenne (1806–1875), who in the 1861 edition of his book Paraplegie hypertrophique de l'enfance de cause cerebrale, described and detailed the case of a boy who had this condition. A year later, he presented photos of his patient in his Album de photographies pathologiques. In 1868, he gave an account of 13 other affected children. Duchenne was the first to do a biopsy to obtain tissue from a living patient for microscopic examination.[65][66]
Notable cases
Alfredo ("Dino", "Alfredino") Ferrari (born January 1932 in Modena), the son of Enzo Ferrari, designed the 1.5 L DOHC V6 engine for the model F2 at the end of 1955. But Dino never saw the engine produced: he died on 30 June 1956 in Modena at the age of 24, before his namesakes Dino and Fiat Dino were made.
Rapper Darius Weems had the disease and used his notoriety to raise awareness and funds for treatment.[67] He died at the age of 27 (his brother also had the disease, until his death at the age of 19). The film Darius Goes West documents Weems's journey of growth and acceptance of having the disease.
Jonathan Evison's novel, The Revised Fundamentals of Caregiving, published in 2012, depicted a young man affected by the disease. In 2016, Netflix released The Fundamentals of Caring, a film based on the novel.[68]
Research
Current research includes exon-skipping, stem cell replacement therapy, analog up-regulation, gene replacement, and supportive care to slow disease progression.
Efforts are ongoing to find medications that either return the ability to make dystrophin or utrophin.[69] Other efforts include trying to block the entry of calcium ions into muscle cells.[70]
Exon-skipping
Antisense oligonucleotides (oligos), structural analogs of DNA, are the basis of a potential treatment for 10% of people with DMD.[71] The compounds allow faulty parts of the dystrophin gene to be skipped when it is transcribed to RNA for protein production, permitting a still-truncated but more functional version of the protein to be produced.[72] It is also known as nonsense suppression therapy.[73]
Two kinds of antisense oligos, 2'-O-methyl phosphorothioate oligos (like drisapersen) and Morpholino oligos (like eteplirsen), have tentative evidence of benefit and are being studied.[74] Eteplirsen is targeted to skip exon 51. "As an example, skipping exon 51 restores the reading frame of ~ 15% of all the boys with deletions. It has been suggested that by having 10 AONs to skip 10 different exons it would be possible to deal with more than 70% of all DMD boys with deletions."[71] This represents about 1.5% of cases.[71]
People with Becker's muscular dystrophy, which is milder than DMD, have a form of dystrophin which is functional even though it is shorter than normal dystrophin.[75] In 1990 England et al. noticed that a patient with mild Becker muscular dystrophy was lacking 46% of his coding region for dystrophin.[75] This functional, yet truncated, form of dystrophin gave rise to the notion that shorter dystrophin can still be therapeutically beneficial. Concurrently, Kole et al. had modified splicing by targeting pre-mRNA with antisense oligonucleotides (AONs).[76] Kole demonstrated success using splice-targeted AONs to correct missplicing in cells removed from beta-thalassemia patients[77][78] Wilton's group tested exon skipping for muscular dystrophy.[79][80]
Gene therapy
Researchers are working on a gene editing method to correct a mutation that leads to Duchenne muscular dystrophy (DMD).[81] Researchers used a technique called CRISPR/Cas9-mediated genome editing, which can precisely remove a mutation in the dystrophin gene in DNA, allowing the body's DNA repair mechanisms to replace it with a normal copy of the gene.[82][83] The benefit of this over other gene therapy techniques is that it can permanently correct the "defect" in a gene rather than just transiently adding a "functional" one.
Genome editing through the CRISPR/Cas9 system is not currently feasible in humans. However, it may be possible, through advancements in technology, to use this technique to develop therapies for DMD in the future.[84][85] In 2007, researchers did the world's first clinical (viral-mediated) gene therapy trial for Duchenne MD.[86]
Biostrophin is a delivery vector for gene therapy in the treatment of Duchenne muscular dystrophy and Becker muscular dystrophy.[87]
References
- "Duchenne". Merriam-Webster Dictionary.
- "NINDS Muscular Dystrophy Information Page". NINDS. March 4, 2016. Archived from the original on 30 July 2016. Retrieved 12 September 2016.
- "Muscular Dystrophy: Hope Through Research". NINDS. March 4, 2016. Archived from the original on 30 September 2016. Retrieved 12 September 2016.
- Lisak RP, Truong DD, Carroll W, Bhidayasiri R (2011). International Neurology. Wiley. p. 222. ISBN 9781444317015.
- Nozoe KT, Akamine RT, Mazzotti DR, Polesel DN, Grossklauss LF, Tufik S (2016). "Phenotypic contrasts of Duchenne Muscular Dystrophy in women: Two case reports". Sleep Sci. 9 (3): 129–133. doi:10.1016/j.slsci.2016.07.004. PMC 5241604. PMID 28123647.
- Hamilton, Jon (27 July 2020). "A Boy with Muscular Dystrophy Was Headed for a Wheelchair. Then Gene Therapy Arrived". NPR.
- "Muscular Dystrophy: Hope Through Research". National Institute of Neurological Disorders and Stroke. Retrieved 10 August 2020.
- "Duchenne muscular dystrophy | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2021-01-24.
- Angelini C, Tasca E (December 2012). "Fatigue in muscular dystrophies". Neuromuscular Disorders. 22 Suppl 3: S214-20. doi:10.1016/j.nmd.2012.10.010. PMC 3526799. PMID 23182642.
- Rowland, L.P. (1985). "Clinical Perspective: Phenotypic Expression In Muscular Dystrophy". In Strohman, C.; Wolf, S. (eds.). Gene Expression in Muscle. Advances in Experimental Medicine and Biology. Plenum Press. pp. 3–5. ISBN 978-1-4684-4907-5.
- Darras BT, Urion DK, Ghosh PS, Adam MP, Ardinger HH, Pagon RA, et al. (2018). "Dystrophinopathies". PMID 20301298.
{{cite journal}}: Cite journal requires|journal=(help) - Emery AE, Muntoni F, Quinlivan RC (2015). Duchenne Muscular Dystrophy (Fourth ed.). OUP Oxford. ISBN 978-0-19968148-8. Retrieved 27 May 2020.
- "Muscular dystrophy - Symptoms and causes". Mayo Clinic. Archived from the original on 2015-02-06. Retrieved 2015-02-06.
- MedlinePlus Encyclopedia: Duchenne muscular dystrophy
- Sutherland DH, Olshen R, Cooper L, Wyatt M, Leach J, Mubarak S, et al. (February 1981). "The pathomechanics of gait in Duchenne muscular dystrophy". Developmental Medicine and Child Neurology. 23 (1): 3–22. doi:10.1111/j.1469-8749.1981.tb08442.x. PMID 7202868. S2CID 895379.
- Baptista CR, Costa AA, Pizzato TM, Souza FB, Mattiello-Sverzut AC (2014). "Postural alignment in children with Duchenne muscular dystrophy and its relationship with balance". Brazilian Journal of Physical Therapy. 18 (2): 119–26. doi:10.1590/s1413-35552012005000152. PMC 4183248. PMID 24838810.
- Doorenweerd N, Mahfouz A, van Putten M, Kaliyaperumal R, T' Hoen P, Hendriksen J, et al. (3 October 2017). "Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy". Scientific Reports. 7 (1): 12575. Bibcode:2017NatSR...712575D. doi:10.1038/s41598-017-12981-5. PMC 5626779. PMID 28974727.
- Online Mendelian Inheritance in Man (OMIM): Muscular Dystrophy, Duchenne Type; DMD - 310200
- Vera, Carlos D.; Zhang, Angela; Pang, Paul D.; Wu, Joseph C. (2022). "Treating Duchenne Muscular Dystrophy: The Promise of Stem Cells, Artificial Intelligence, and Multi-Omics". Frontiers in Cardiovascular Medicine. 9: 851491. doi:10.3389/fcvm.2022.851491. ISSN 2297-055X. PMC 8960141. PMID 35360042.
- "Duchenne Muscular Dystrophy: Pathophysiological Implications of Mitochondrial Calcium Signaling and ROS Production". 2012-05-02. Archived from the original on May 2, 2012. Retrieved 2014-06-29.
- Wahl, Margaret (October 21, 2016). "Quest - Article - But Girls Don't Get Duchenne, or Do They? - A Quest Article". Muscular Dystrophy Association. Retrieved July 6, 2019.
- "Understanding Genetics". 26 November 2021.
- Nico B, Ribatti D (January 2012). "Morphofunctional aspects of the blood-brain barrier". Current Drug Metabolism. 13 (1): 50–60. doi:10.2174/138920012798356970. PMID 22292807.
- "University of Utah Muscular Dystrophy". Genome.utah.edu. 2009-11-28. Retrieved 2013-02-16.
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. (January 2010). "Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management". The Lancet. Neurology. 9 (1): 77–93. CiteSeerX 10.1.1.176.4466. doi:10.1016/s1474-4422(09)70271-6. PMID 19945913. S2CID 328499.
- Nicholson LV, Johnson MA, Bushby KM, Gardner-Medwin D, Curtis A, Ginjaar IB, den Dunnen JT, Welch JL, Butler TJ, Bakker E (September 1993). "Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 2. Correlations within individual patients". Journal of Medical Genetics. 30 (9): 737–44. doi:10.1136/jmg.30.9.737. PMC 1016530. PMID 8411068.
- Muntoni F (August 2001). "Is a muscle biopsy in Duchenne dystrophy really necessary?". Neurology. 57 (4): 574–5. doi:10.1212/wnl.57.4.574. PMID 11524463. S2CID 13474827.
- Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB (April 2003). "Rapid direct sequence analysis of the dystrophin gene". American Journal of Human Genetics. 72 (4): 931–9. doi:10.1086/374176. PMC 1180355. PMID 12632325.
- Beksac MS, Tanacan A, Aydin Hakli D, Orgul G, Soyak B, Balci Hayta B, et al. (30 July 2018). "Gestational Outcomes of Pregnant Women Who Have Had Invasive Prenatal Testing for the Prenatal Diagnosis of Duchenne Muscular Dystrophy". Journal of Pregnancy. 2018: 1–5. doi:10.1155/2018/9718316. PMC 6091284. PMID 30151283.
- Devaney, Stephanie A.; Palomaki, Glenn E.; Scott, Joan A.; Bianchi, Diana W. (2011-08-10). "Noninvasive Fetal Sex Determination Using Cell-Free Fetal DNA: A Systematic Review and Meta-analysis". JAMA. 306 (6): 627–636. doi:10.1001/jama.2011.1114. ISSN 0098-7484. PMC 4526182. PMID 21828326.
- "Duchenne Muscular Dystrophy Statement". Drug Safety and Availability. US FDA. 2014-10-31. Archived from the original on 2014-11-02.
- "Pfizer's New Phase 1b Results of Gene Therapy in Ambulatory Boys with Duchenne Muscular Dystrophy (DMD) Support Advancement into Pivotal Phase 3 Study | Pfizer".
- Dany A, Barbe C, Rapin A, Réveillère C, Hardouin JB, Morrone I, et al. (November 2015). "Construction of a Quality of Life Questionnaire for slowly progressive neuromuscular disease". Quality of Life Research. 24 (11): 2615–23. doi:10.1007/s11136-015-1013-8. PMID 26141500. S2CID 25834947.
- Falzarano MS, Scotton C, Passarelli C, Ferlini A (October 2015). "Duchenne Muscular Dystrophy: From Diagnosis to Therapy". Molecules. 20 (10): 18168–84. doi:10.3390/molecules201018168. PMC 6332113. PMID 26457695.
- Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY (May 2016). "Corticosteroids for the treatment of Duchenne muscular dystrophy". The Cochrane Database of Systematic Reviews. 5 (5): CD003725. doi:10.1002/14651858.CD003725.pub4. PMC 8580515. PMID 27149418.
- Verhaert D, Richards K, Rafael-Fortney JA, Raman SV (January 2011). "Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations". Circulation: Cardiovascular Imaging. 4 (1): 67–76. doi:10.1161/CIRCIMAGING.110.960740. PMC 3057042. PMID 21245364.
- "Railroading at the FDA". Nature Biotechnology. 34 (11): 1078. 2016-11-08. doi:10.1038/nbt.3733. PMID 27824847.
- "FDAetep". Food and Drug Administration. 19 September 2016. Retrieved 8 July 2019.
- "CHMP Advises Against Approval for Eteplirsen in DMD". Medscape. Retrieved 2019-07-09.
- "Translarna EPAR". European Medicines Agency (EMA). Retrieved 14 August 2020.
- "Translarna - Summary of Product Characteristics (SmPC)". (emc). 24 April 2017. Retrieved 18 June 2017.
- "FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation". U.S. Food and Drug Administration (FDA) (Press release). 12 December 2019. Archived from the original on 13 December 2019. Retrieved 12 December 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - "Drug Approval Package: Vyondys 53 (golodirsen)". U.S. Food and Drug Administration (FDA). 21 January 2020. Retrieved 22 January 2020.
- "FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation". U.S. Food and Drug Administration (FDA) (Press release). 12 August 2020. Retrieved 12 August 2020. This article incorporates text from this source, which is in the public domain.
- "FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation". U.S. Food and Drug Administration (FDA) (Press release). 25 February 2021. Retrieved 25 February 2021. This article incorporates text from this source, which is in the public domain.
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. (March 2018). "Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management". Lancet Neurol. 17 (3): 251–267. doi:10.1016/S1474-4422(18)30024-3. PMC 5869704. PMID 29395989.
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. (April 2018). "Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management". Lancet Neurol. 17 (4): 347–361. doi:10.1016/S1474-4422(18)30025-5. PMC 5889091. PMID 29395990.
- "Duchenne Muscular Dystrophy". Physiopedia. Retrieved 2022-10-10.
- Pedlow, Katy; McDonough, Suzanne; Lennon, Sheila; Kerr, Claire; Bradbury, Ian (2019-10-13). "Assisted standing for Duchenne muscular dystrophy". The Cochrane Database of Systematic Reviews. 2019 (10): CD011550. doi:10.1002/14651858.CD011550.pub2. ISSN 1469-493X. PMC 6790222. PMID 31606891.
- "Muscular Dystrophy Information Page: National Institute of Neurological Disorders and Stroke (NINDS)". www.ninds.nih.gov. Archived from the original on 30 July 2016. Retrieved 22 February 2022.
- "Muscular Dystrophy: Hope Through Research". www.ninds.nih.gov. Archived from the original on 30 September 2016. Retrieved 22 February 2022.
- "NINDS Muscular Dystrophy Information Page". NINDS. March 4, 2016. Archived from the original on 30 July 2016. Retrieved 12 September 2016.
- "Duchenne muscular dystrophy (DMD) | Muscular Dystrophy Campaign". Muscular-dystrophy.org. Archived from the original on 2013-01-21. Retrieved 2013-02-16.
- Carter, Nicole (January 14, 2021). "Nursing home resident defies COVID, wants to eat out again". Sun Journal.
- Kieny P, Chollet S, Delalande P, Le Fort M, Magot A, Pereon Y, et al. (September 2013). "Evolution of life expectancy of patients with Duchenne muscular dystrophy at AFM Yolaine de Kepper centre between 1981 and 2011". Annals of Physical and Rehabilitation Medicine. 56 (6): 443–54. doi:10.1016/j.rehab.2013.06.002. PMID 23876223.
- Krajina A, Podrabský P, Steinhart L, Endrys J, Coufal L (2012-11-22). "[Personal experimental experience with the administration of liquid obliterative agents using percutaneous intra-arterial balloon catheters with a controlled leak]". Sbornik Vedeckych Praci Lekarske Fakulty Karlovy Univerzity V Hradci Kralove. Supplementum. 30 (2): 201–11. doi:10.1186/1750-1172-7-S2-A8. PMC 3504593. PMID 3504593.
- Marques MJ, Ferretti R, Vomero VU, Minatel E, Neto HS (March 2007). "Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy". Muscle & Nerve. 35 (3): 349–53. doi:10.1002/mus.20697. PMID 17143878. S2CID 41968787.
- Ferretti R, Marques MJ, Khurana TS, Santo Neto H (June 2015). "Expression of calcium-buffering proteins in rat intrinsic laryngeal muscles". Physiological Reports. 3 (6): e12409. doi:10.14814/phy2.12409. PMC 4510619. PMID 26109185.
- Feng X, Files DC, Zhang T (2014). "Intrinsic Laryngeal Muscles and Potential Treatments for Skeletal Muscle-Wasting Disorders". Austin Journal of Otolaryngology. 1 (1): 3. Archived from the original on 2015-06-26.
- White Z, Hakim CH, Theret M, Yang NN, Rossi F, Cox D, Francis GA, Straub V, Selby K, Panagiotopoulos C, Duan D, Bernatchez P (July 2020). "High prevalence of plasma lipid abnormalities in human and canine Duchenne and Becker muscular dystrophies depicts a new type of primary genetic dyslipidemia". J Clin Lipidol. 14 (4): 459–69. doi:10.1016/j.jacl.2020.05.098. PMC 7492428. PMID 32593511. S2CID 219741257.
- "Key Findings: Prevalence of Duchenne / Becker Muscular Dystrophies". Centers for Disease Control and Prevention. 2018-01-05. Retrieved 2018-11-18.
- Politano L. "Cardiomiologia e Genetica Medica" [Cardiomyology and Medical Genetics] (in Italian). Seconda Università degli Studi di Napoli. Archived from the original on July 4, 2015. Retrieved August 24, 2015.
- De Rosa G (October 2005). "Da Conte a Duchenne" [By Conte in Duchenne]. DM (in Italian). Unione Italiana Lotta alla Distrofia Muscolare. Archived from the original on March 4, 2016. Retrieved August 24, 2015.
- Nigro G (December 2010). "One-hundred-seventy-five years of Neapolitan contributions to the fight against the muscular diseases". Acta Myologica. 29 (3): 369–91. PMC 3146338. PMID 21574522.
- "Duchenne muscular dystrophy". Medterms.com. 2011-04-27. Archived from the original on 2012-08-06. Retrieved 2013-02-16.
- Duchenne de Boulogne at Who Named It?
- McFadden C (November 22, 2012). "Darius Weems' Next Chapter: Rap Star With Duchenne Muscular Dystrophy Tries Clinical Trial". ABC News. Archived from the original on August 5, 2016. Retrieved June 29, 2016.
- Berkshire, Geoff (23 January 2016). "Sundance Film Review: 'The Fundamentals of Caring'". Variety. Retrieved 21 October 2021.
- Guiraud S, Davies KE (June 2017). "Pharmacological advances for treatment in Duchenne muscular dystrophy". Current Opinion in Pharmacology. 34: 36–48. doi:10.1016/j.coph.2017.04.002. PMID 28486179.
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP (February 2010). "Calcium and the damage pathways in muscular dystrophy". Canadian Journal of Physiology and Pharmacology. 88 (2): 83–91. doi:10.1139/Y09-058. PMID 20237582.
- Scoto M, Finkel R, Mercuri E, Muntoni F (August 2018). "Genetic therapies for inherited neuromuscular disorders" (PDF). The Lancet. Child & Adolescent Health. 2 (8): 600–609. doi:10.1016/S2352-4642(18)30140-8. PMID 30119719. S2CID 52032568.
- Dunckley MG, Manoharan M, Villiet P, Eperon IC, Dickson G (July 1998). "Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides". Human Molecular Genetics. 7 (7): 1083–90. doi:10.1093/hmg/7.7.1083. PMID 9618164.
- Finkel RS (September 2010). "Read-through strategies for suppression of nonsense mutations in Duchenne/ Becker muscular dystrophy: aminoglycosides and ataluren (PTC124)". Journal of Child Neurology. 25 (9): 1158–64. doi:10.1177/0883073810371129. PMC 3674569. PMID 20519671.
- "FDA grants accelerated approval to first drug for Duchenne muscular dystrophy" (Press release). FDA. September 19, 2016. Archived from the original on December 11, 2016. Retrieved 2016-12-12.
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE, Bulman DE, Harris JB, Davies KE (January 1990). "Very mild muscular dystrophy associated with the deletion of 46% of dystrophin". Nature. 343 (6254): 180–2. Bibcode:1990Natur.343..180E. doi:10.1038/343180a0. PMID 2404210. S2CID 4349360.
- Dominski Z, Kole R (September 1993). "Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides". Proceedings of the National Academy of Sciences of the United States of America. 90 (18): 8673–7. Bibcode:1993PNAS...90.8673D. doi:10.1073/pnas.90.18.8673. PMC 47420. PMID 8378346.
- Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, Kole R (August 2000). "Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients". Proceedings of the National Academy of Sciences of the United States of America. 97 (17): 9591–6. Bibcode:2000PNAS...97.9591L. doi:10.1073/pnas.97.17.9591. PMC 16909. PMID 10944225.
- Suwanmanee T, Sierakowska H, Lacerra G, Svasti S, Kirby S, Walsh CE, Fucharoen S, Kole R (September 2002). "Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides". Molecular Pharmacology. 62 (3): 545–53. doi:10.1124/mol.62.3.545. PMID 12181431.
- Wilton SD, Lloyd F, Carville K, Fletcher S, Honeyman K, Agrawal S, Kole R (July 1999). "Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides". Neuromuscular Disorders. 9 (5): 330–8. doi:10.1016/S0960-8966(99)00010-3. PMID 10407856. S2CID 20678312.
- Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S (July 2007). "Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript". Molecular Therapy. 15 (7): 1288–96. doi:10.1038/sj.mt.6300095. PMID 17285139.
- Long C, Li H, Tiburcy M, Rodriguez-Caycedo C, Kyrychenko V, Zhou H, Zhang Y, Min YL, Shelton JM, Mammen PP, Liaw NY, Zimmermann WH, Bassel-Duby R, Schneider JW, Olson EN (January 2018). "Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing". Science Advances. 4 (1): eaap9004. Bibcode:2018SciA....4P9004L. doi:10.1126/sciadv.aap9004. PMC 5796795. PMID 29404407.
- Cohen J (2018-08-30). "Gene editing of dogs offers hope for treating human muscular dystrophy". Science. doi:10.1126/science.aav2676. S2CID 92204241.
- Patmanathan SN, Gnanasegaran N, Lim MN, Husaini R, Fakiruddin KS, Zakaria Z (2018). "CRISPR/Cas9 in Stem Cell Research: Current Application and Future Perspective". Current Stem Cell Research & Therapy. 13 (8): 632–644. doi:10.2174/1574888X13666180613081443. PMID 29895256. S2CID 48357156.
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN (September 2014). "Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA". Science. 345 (6201): 1184–1188. Bibcode:2014Sci...345.1184L. doi:10.1126/science.1254445. PMC 4398027. PMID 25123483.
- Wade N (31 December 2015). "Gene Editing Offers Hope for Treating Duchenne Muscular Dystrophy, Studies Find". The New York Times. Archived from the original on 2 January 2016. Retrieved 1 January 2016.
- Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR (September 2007). "Gene therapy for duchenne muscular dystrophy: expectations and challenges". Archives of Neurology. 64 (9): 1236–41. doi:10.1001/archneur.64.9.1236. PMID 17846262.
- Khurdayan VK, Bozzo J, Prous JR (October 2005). "Chronicles in drug discovery". Drug News & Perspectives. 18 (8): 517–22. doi:10.1358/dnp.2005.18.8.953409. PMID 16391721.
Further reading
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. (March 2018). "Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management". Lancet Neurol. 17 (3): 251–267. doi:10.1016/S1474-4422(18)30024-3. PMC 5869704. PMID 29395989.
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. (April 2018). "Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management". Lancet Neurol. 17 (4): 347–361. doi:10.1016/S1474-4422(18)30025-5. PMC 5889091. PMID 29395990.
External links
- Muscular Dystrophies at Curlie
- CDC's National Center on Birth Defects and Developmental Disabilities (previously listed below as "Duchenne/Becker Muscular Dystrophy, NCBDDD") at CDC
- Genes and Disease Page at NCBI
| Types | |
|---|---|
| National/International Organizations |
|
| National/International Events |
|
| Clinical trials | |