Remodelación de la cromatina
La remodelación de la cromatina es la modificación dinámica de la arquitectura de la cromatina para permitir el acceso de proteínas reguladoras de la transcripción al ADN genómico empaquetado, controlando así la expresión génica. Esta remodelación es realizada principalmente por: 1) Modificaciones covalentes de las histonas por enzimas específicas (ej.: histona acetiltransferesas (HATs), deacetilasas, metiltransferasas y quinasas; y 2) Complejos proteicos remodeladores de la cromatina dependientes de ATP, los cuales mueven, eyectan o restructuran los nucleosomas.[1] Además de regular activamente la expresión génica, la remodelación dinámica de la cromatina cumple un rol regulador epigenético para diferentes procesos biológicos, tales como la replicación y reparación del ADN, apoptosis, segregación de cromosomas, así como el desarrollo celular y la pluripotencia. Las alteraciones en proteínas remodeladoras de la cromatina se asocian con enfermedades humanas como el cáncer, por lo que, actualmente, se estudian rutas implicadas en remodelación de la cromatina en búsqueda de dianas para estrategias terapéuticas de tratamiento del cáncer.
Introducción
La regulación transcripcional en el genoma se controla principalmente, en un estadio inicial, por la unión de proteínas reguladoras (ej.: ARN polimerasa, factores de transcripción y activadores y represoras de la transcripción) a la secuencia promotora de la región codificante del ADN. Sin embargo, el ADN se encuentra densamente empaquetado en el núcleo celular con la colaboración de proteínas de empaquetamiento, principalmente histonas que forman unidades repetidas de nucleosomas, los cuales se agrupan formando la estructura condensada de la cromatina. Esta condensación oculta gran cantidad de regiones del ADN a la acción de proteínas reguladoras de la expresión génica. El proceso de la remodelación de la cromatina, al modificar la arquitectura de los nucleosomas y exponer u ocultar regiones del ADN, permite superar este obstáculo y permitir el acceso dinámico al ADN para la regulación de la transcripción.
Por definición, la remodelación de la cromatina es un proceso mediado por enzimas para facilitar el acceso de los nucleosomas al modificar la estructura, composición y posicionamiento de estos.
Clasificación
El acceso de los nucleosomas al ADN está controlado principalmente por dos clases de complejos proteicos:
- Complejos modificadores de histonas (modificaciones covalentes).
- Complejos remodeladores de la cromatina dependientes de ATP.
Complejos modificadores de histonas
Determinados complejos proteicos catalizan adición o eliminación de componentes químicos en las histonas. Estas modificaciones enzimáticas incluyen acetilación, metilación, fosforilación y ubiquitinización, ocurriendo principalmente en los extremos N-terminal de las histonas. Estas afectan a la afinidad de unión de las histonas al ADN, relajando o fortaleciendo la condensación del ADN sobre las histonas, por ejemplo, la metilación de determinados residuos de lisinas en las histonas H3 y H4 genera una mayor condensación del ADN, previniendo así la unión de factores de transcripción que active la expresión génica. Por el contrario, la acetilación de histonas reduce la condensación de la cromatina y expone el ADN a factores de transcripción, conllevando un incremento de la expresión génica.[2]
Modificaciones conocidas
Entre las modificaciones de histonas mejor estudiadas se encuentran:[3]
Los residuos de lisina y arginina se pueden metilar. Las lisinas metiladas son las marcas o modificaciones mejor conocidas del código de las histonas, ya que estas se corresponden con un estado de expresión génica activa. La metilación de lisinas H3K4 y H3K36 se correlacionan con activación transcripcional, mientras que la desmetilación de H3K4 se correlaciona con el silenciamiento de esta. La metilación de lisinas H3K9 y H3K27 se correlaciona con represión transcripcional.[4] La histona H3K9me3 en concreto está altamente relacionada con la heterocromatina constitutiva.[5]
- Acetilación - por HAT (histona acetiltransferasas); desacetilación - por HDAC (histona deacetilasas)
La acetilación suele caracterizar a las regiones abiertas de la cromatina, ya que las histonas acetiladas no se pueden empaquetar tan bien como las desacetiladas.
Existen muchas más modificaciones de histonas, cuyo catálogo se ha expandido gracias a los métodos de espectrometría de masas.[6]
Hipótesis del código de histonas
El código de histonas es una hipótesis que plantea que la transcripción de la información genética codificada en el ADN está en parte regulada por modificaciones químicas de las histonas, principalmente en sus extremos N-terminal. Además de otras modificaciones, como la metilación del ADN, estas constituyen el código epigenético.
Existe evidencia de que este código está escrito por enzimas específicas, las cuales pueden, por ejemplo, metilar o acetilar el ADN ("escritoras"), eliminado por otras enzimas con actividad demetilasa o deacetilasa ("borradoras"), y finalmente, identificado por proteínas ("lectoras"), las cuales son reclutadas a estas modificaciones uniéndose vía dominios específicos (ej.: bromodominio, cromodominio etc.). Esta triple acción de "escribir", "leer" y "borrar" establecen un ambiente local favorable para la regulación transcripcional, reparación del ADN etc.[7]
El concepto crítico de la hipótesis de código de histonas es que las modificaciones sirven para reclutar otras proteínas mediante reconocimiento específico de la histona por los dominios especializados de las proteínas, en lugar de simplemente estabilizar y desestabilizar la interacción histona-ADN. Estas proteínas modifican la estructura de la cromatina de manera activa.
A continuación se presenta un resumen del código de histonas y su relación con el estado de la expresión génica (nomenclatura explicada aquí):
| Tipo de modificación | Histona | ||||||
|---|---|---|---|---|---|---|---|
| H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H4K20 | H2BK5 | |
| monometilación | activación[8] | activación[9] | activación[9] | activación[9][10] | activación[9] | activación[9] | |
| dimetilación | represión[4] | represión[4] | activación[10] | ||||
| trimetilación | activación[11] | represión[9] | represión[9] | activación,[10] represión[9] |
represión[4] | ||
| acetilación | activación[11] | activación[11] | |||||
Remodelación de la cromatina dependiente de ATP
Los complejos remodeladores de cromatina dependientes de ATP regulan la expresión génica al mover, eyectar o restructurar los nucleosomas. Estos complejos proteicos tienen un dominio común de actividad ATPasa, el cual, mediante la energía obtenida al hidrolizar ATP, permite reposicionar nucleosomas (proceso usualmente denominado "deslizamiento de nucleosoma"), eyectar o ensamblar histonas o facilitar el intercambio de variantes de histonas, creando así regiones libres de nucleosomas.[12] También, diferentes remodeladores son capaces de translocar el ADN para realizar remodelaciones específicas.[13]
Todos los complejos tienen una subunidad ATPasa, la cual pertenece a la superfamilia de proteínas SNF2. Se han identificado dos grupos principales de subunidades implicadas, conocidos como el grupo SWI2/SNF2 y el grupo de imitación ISWI. Recientemente se ha identificado complejos dependientes de ATP que contienen una subunidad similar a Snf2 ATPasa, la cual también tiene actividad deacetilasa.[14]
Complejos remodeladores de la cromatina conocidos
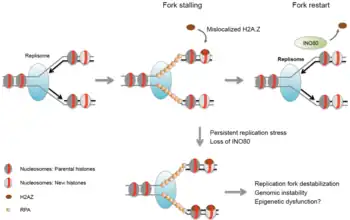
Existen al menos 4 familias de remodeladores de la cromatina en eucariotas: SWI/SNF, ISWI, NuRD/Mi-2/CHD y INO80. Las dos primeras son las mejor estudiadas en la actualidad, especialmente en levaduras. Aunque todos los remodeladores comparten un dominio común ATPasa, cumplen funciones específicas para diferentes procesos biológicos (reparación del ADN, apoptosis etc.) Esto se debe al hecho de que cada complejo tiene dominios proteicos únicos (helicasa, bromodominio etc.) en su región ATPasa, además de que están compuestas de diferentes subunidades.
Funciones específicas
- Múltiples experimentos in vitro sugieren que los remodeladores ISWI organizan los nucleosomas en grupos, creando espacios igualados entre estos, mientras que los remodeladores SWI/SNF desordenan los nucleosomas.
- Se ha comprobado que los remodeladores ISWI cumplen un rol central en el empaquetamiento de la cromatina y el mantenimiento de su estructura tras la replicación del ADN.
- Los remodeladores SWI/SNF e INO80 participan en la reparación de roturas de doble cadena en el ADN (DSB) y de pérdida de nucleótidos (NER), por lo que cumplen un papel importante en la respuesta a daños en el ADN mediada por TP53.
- Los remodeladores NuRD/Mi-2/CHD participan principalmente en la represión de la transcripción y son necesarios para el mantenimiento de la pluripotencia en células madre embrionarias.[12]
Relevancia
En procesos biológicos (condiciones normales)
La remodelación de la cromatina cumple un rol central en la regulación de la expresión génica al permitir el acceso al genoma desempaquetado de la maquinaria transcripcional. Además, el desplazamiento de los nucleosomas por los remodeladores es esencial para diversos procesos biológicos: condensar y segregar cromosomas, replicación y reparación del ADN, desarrollo embrionario, pluripotencia y progresión del ciclo celular. La desregulación de los remodeladores de la cromatina causa la pérdida de la regulación transcripcional en los puntos de control críticos para las funciones biológicas, causando así diferentes enfermedades, como el cáncer.
Respuesta al daño del ADN
La relajación de la cromatina es una de las respuestas más tempranas a cualquier daño en el ADN.[15] Se han llevado a cabo múltiples experimentos sobre la cinética de reclutamiento de proteínas implicadas en la respuesta a daños en el ADN.[16] Esta parece iniciarse por PARP1, cuya acumulación en el sitio de reparación alcanza el 50% en 1,6 segundos tras la rotura del ADN. Rápidamente ocurre la acumulación del remodelador Alc1, el cual tiene un dominio de unión a ADP-ribosa, permitiéndole unirse rápidamente al producto de PARP1. La máxima acumulación de Alc1 ocurre 10 segundos después de la rotura del ADN.[15] En este tiempo ocurre la relajación de la cromatina.[15] La acción de PARP1 permite el reclutamiento de enzimas de reparación MRE11 (50% de acumulación en 13 segundos) y NBS1 (50% de acumulación en 28 segundos).[16]
Otra ruta de relajación de la cromatina, tras la rotura de la doble cadena de ADN, incluye a γ-H2AX, la forma fosforilada de la proteína H2AX. La histona H2AX constituye aproximadamente el 10% de las histonas H2A en la cromatina humana.[17] La γ-H2AX (fosforilada en la serina 139 de H2AX) se detectó 20 segundos y alcanzó 50% de acumulación 1 minuto después de la irradiación de las células, agente causante de la rotura de doble cadena de ADN.[17] En la región de reparación, la extensión de cromatina con γ-H2AX fosforilada era de 2 Mbs.[17]
γ-H2AX no causa condensación de la cromatina por sí misma, aunque, tras pocos segundos de radiación, la proteína MDC1 ("Mediador de control de daño en el ADN 1") sí se une específicamente a γ-H2AX.[18][19] A esto se suma una acumulación lineal de la proteína RNF8 y la proteína de reparación NBS1, la cual se une a MDC1, conforme esta se une a gamma-H2AX.[20] RNF8 colabora en el desempaquetamiento de la cromatina, vía interacción con la proteína CHD4,[21] un componente del complejo desacetilasa NuRD, que también es remodelador de la cromatina. La acumulación de CHD4 en el ADN dañado es rápida, alcanzando un 50% de acumulación 40 segundos tras la radiación..[22]
Tras la rápida relajación inicial de la cromatina en el lugar de daño en el ADN, ocurre una re-condensación rápida, devolviendo la cromatina a un estado de empaquetamiento similar al estado anterior al daño causado, todo en aproximadamente en 20 minutos.[15]
Cáncer
La remodelación de la cromatina tiene un papel regulador en momentos cruciales del crecimiento y división celular, como la progresión del ciclo celular, reparación del ADN y segregación de los cromosomas. Por lo tanto, tiene un papel supresor de tumores. Las mutaciones en estos remodeladores de la cromatina y las modificaciones covalentes en histonas pueden favorecer potencialmente la autosuficiencia en el crecimiento celular y el escape de las señales celulares reguladoras del crecimiento, dos importantes características del cáncer.[23]
- Se han identificado mutaciones de inactivación de la proteína SMARCB1, anteriormente denominada hSNF5/INI1, en tumores rabdoides, los cuales afectar comúnmente a población pediátrica.[24] Esta proteína es un componente del complejo remodelador SWI/SNF en humanos. También existen mutaciones similares en otros cánceres pediátricos, como el carcinoma de plexo coroideo, meduloblastoma y en algunas leucemias agudas. Además, estudios de silenciamiento de genes en ratones indican que SMARCB1 es un supresor de tumores. Se han identificado más subunidades del remodelador SWI/SNF humano con mutaciones presentes en tumores.[25]
- La ATPasa BRG1, componente de SWI/SNF (también llamada SMARCA4) es la ATPasa remodeladora de la cromatina que presenta mutaciones más frecuentemente en cáncer.[26] Las mutaciones de este gen se identificaron por primera vez en líneas celulares de cáncer humanas extraídas de glándula adrenal[27] y el pulmón.[28] En cáncer las mutaciones en BRG1 muestran una preferencia inusualmente altas por mutaciones sin sentido en secuencias conservadas del dominio ATPasa,[29][26] alterando partes funcionales importantes para el bolsillo de captación de ATP o la unión al ADN.[30][29] Se ha comprobado que estas mutaciones son genéticamente dominantes, alterando la función reguladoras de la cromatina en enhancers[29] y promotores.[30]
- La fusión de proteínas PML-RARA en leucemia mieloide aguda recluta histona deacetilasas. Esto genera una represión de los genes responsables de la diferenciación de los mielocitos y esto a su vez genera leucemia.[31]
- La proteína supresora de tumores Rb recluta a las proteínas homólogas en humanos de las enzimas de la familia SWI/SNF BRG1, histona deacetilasa y ADN metiltransferasa. Las mutaciones en BRG1 se han identificado en diferentes cánceres, generando pérdida de la acción supresora de tumores de Rb.[32]
- Investigaciones recientes han identificado hipermetilación del ADN en la región promotora de genes supresores de tumores en diferentes tipos de cáncer. Aunque se han encontrado pocas mutaciones aún en histona metiltransferasas,se ha comprobado correlación entre la hipermetilación del ADN y la metilación de histona H3 en la lisina 9 (H3K9me) en cáncer, sobre todo en cáncer colorrectal y de mama.
- Las mutaciones en histona acetiltransferasas (HAT) p300, las cuales provocan proteínas truncadas o mutaciones sin sentido son comúnmente identificadas en cáncer colorrectal, de páncreas, de mama y gástrico. Un número importante de los casos de glioblastomas presentan pérdida de heterozigosidad en regiones codificantes de p300 (cromosoma 22q13).
- Además, las HATs tienen diferentes roles como factores de transcripción además de tener actividad histona acetilasa, por ejemplo, la subunidad de HAT: hADA3 puede actuar como una proteína adaptadora que se une a factores de transcripción con otros complejos HAT. En ausencia de hADA3, la actividad transcripcional de TP53 se ve significativamente reducida, sugiriendo el rol de hADA3 en activar a TP53 en respuesta a daño en el ADN.
- De manera similar, se ha comprobado que TRRAP, la proteína homóloga humana a Tra1 en levaduras, interactúa directamente con c-Myc y E2F1, dos conocidas oncoproteínas.[33]
Genómica del cáncer
Los rápidos avances en el campo de la genómica del cáncer y en métodos de secuenciación de bisulfito, ChIP-chip y ChIP-Seq están contribuyendo en esclarecer el rol de los remodeladores de la cromatina en la regulación transcripcional y en el cáncer.
Intervención terapéutica
La inestabilidad epigenética causada por la desregulación en los remodeladores de la cromatina se ha estudiado en diferentes tipos de cáncer, incluyendo cáncer de mama, colorrectal, pancreático. Esta inestabilidad causa el silenciamiento de variedad de genes con un impacto directo en genes supresores de tumores. Por lo tanto, se diseñan estrategias para solucionar el silenciamiento epigenético con combinaciones sinergísticas de inhibidores de HDAC o HDI y agentes desmetiladores del ADN. Los HDIs se utilizan principalmente para terapia adyuvante en diferentes tipos de cáncer.[34][35] Los inhibidores de HDAC pueden inducir expresión de p21 (WAF1), un regulador de la actividad supresora de tumores de p53. Los HDACs están involucrados en la ruta de supresión de la proliferación celular de la proteína del retinoblastoma Rb.[36] El estrógeno está bien caracterizado como un factor mitogénico en la tumorogénesis y progresión del cáncer de mama vía la unión al receptor alfa del estrógeno (ER-α). Recientes investigaciones indican que la inactivación de la cromatina por HDAC y metilación del ADN es un componente crítico en el silenciamiento de ER-α en células humanas de cáncer de mama.[37]
- Uso aprobado:
- Vorinostat fue aprobado por la Administración de Alimentos y Medicamentos (FDA) de los EE.UU. en octubre de 2006 para el tratamiento del linfoma cutáneo de células T.[38][39]
- Romidepsin (nombre comercial Istodax) fue aprobado por la FDA de los EE.UU. en noviembre de 2009 para tratar el linfoma cutáneo de células T.[40][41]
- Ensayos clínicos de fase III:
- Panobinostat (LBH589) está en ensayos clínicos para diferentes tipos de cáncer, incluyendo el linfoma cutáneo de células T.
- Ácido valproico (en forma de valproato de magnesio) está en fase III para cáncer de cérvix y de ovarios.
- Inicio de ensayos clínicos pivotales de fase II:
- Belinostat (PXD101) ha pasado los ensayos clínicos de fase II para la recidiva de cáncer de ovarios, y buenos resultados contra el linfoma de células T.
- Inhibidores de HDAC .
Los principales candidatos actuales como dianas terapéuticas son las histona lisina metiltransferasas (KMT) y las arginina metiltransferasas (PRMT).[42]
Otros síndromes de enfermedades
- El síndrome de α-talasemia con retraso mental ligado al cromosoma X (ATR-X) y el síndrome de α-talasemia y mielodisplasia están causados por mutaciones en el gen ATRX, una ATPasa relacionada con SNF2 y que contiene un dominio dedo de zinc PHD.[43]
- El síndrome CHARGE, una enfermedad autosómica dominante, se ha asociado recientemente con la haploinsuficiencia del gen CHD7, que codifica la ATPasa homónima, miembro de la familia de proteínas CHD.[44]
Senescencia
La remodelación de la cromatina está involucrada en el proceso de la senescencia celular, la cual se relaciona con el envejecimiento de un organismo. La senescencia celular se refiere al término permanente del ciclo celular, donde las células, tras la mitosis, siguen siendo metabólicamente activas pero dejan de proliferar[45][46] La senescencia puede surgir debido a la degradación asociada con la edad, el desgaste de los telómeros, progeria, cáncer y otras formas de daño o enfermedad. Las células en este estado sufren diferentes cambios fenotípicos, potencialmente para prevenir la proliferación de células dañadas o cancerosas, con una organización de la cromatina modificada, alteraciones en la abundancia de remodeladores y cambios en las modificaciones epigenéticas.[47][48][45] Ocurre modificaciones en la arquitectura de la cromatina, ya que la heterocromatina constitutiva migra al centro del núcleo y desplaza a la eucromatina y la heterocromatina facultativa a regiones periféricas de este. Esto altera las interacciones cromatina-lámina e invierte el patrón típico de una célula mitoticamente activa.[49][47] Los Dominios asociados a Lámina (LADs) y los topológicamente asociados (TADs) se ven alterados por esta migración, la cual puede afectar a las interacciones cis en el genoma.[50] Adicionalmente, existe un patrón general de pérdida de histonas canónicas, concretamente las histonas H3 y H4, y la histona ligadora H1.[49] Las variantes de histonas con dos exones se encuentran sobre-expresadas en células senescentes, produciendo nucleosomas modificados que contribuyen a una cromatina más permisiva a los cambios en la senescencia.[50] Aunque la transcripción de variantes de histonas puede ser elevada, las histonas canónicas no se expresan debido a que esto solo ocurre en la fase S del ciclo celular y las células senescentes son postmitóticas.[49] Durante la senescencia, partes de los cromosomas pueden ser exportados del núcleo para sufrir degradación por lisosomas, causando un desajuste y alteraciones mayores de la organización e interacciones de la cromatina.[48]
La abundancia de remodeladores de la cromatina puede estar involucrada en la senescencia celular ya que el silenciamiento o bloqueo de estos, tales como NuRD, ACF1 y SWI/SNP, puede resultar en daños en el ADN y fenotipos senescentes en levaduras, C. elegans, ratones y células humanas.[51][48][52] ACF1 y NuRD están infra-expresadas en células senescentes, lo que sugiere que la remodelación de la cromatina es esencial para el mantenimiento del fenotipo mitótico.[51][52] Los genes involucrados en la señalización celular que lleva a la senescencia pueden silenciarse por la conformación de la cromatina y la acción de complejos represores Polycomb, tales como PRC1/PRC2 con p16.[53][54] La eliminación de remodeladores específicos resulta en la inactivación de genes proliferadores al fallar el mantenimiento del silenciamiento.[48] Algunos remodeladores actúan en regiones de enhancers antes que en loci específicos para prevenir la reentrada en el ciclo celular al formar regiones de heterocromatina densa en regiones reguladoras.[54]
Las células en senescencia experimentan extensas modificaciones epigenéticas en regiones concretas de la cromatina, en comparación con células mitóticas. Cuando células humanas y murinas entran en senescencia experimentan un descenso general de los niveles de metilación. Sin embargo, ciertos loci específicos pueden escapar de esta tendencia general.[55][50][48][53] Regiones concretas de la cromatina, especialmente aquellas en torno a promotores y enhancers de loci proliferadores, pueden mostrar niveles altos de metilación con un desequilibrio general de las modificaciones activadoras y represoras de histonas.[47] Los genes proliferadores pueden mostrar un aumento de marcas de unión de la histona represora H3K27me3, mientras que genes involucrados en el silenciamiento o histonas aberrantes pueden encontrarse enriquecidos con la histona activadora H3K4me3.[50] Adicionalmente, las histonas deacetilasas sobre-expresadas, como miembros de la familia de la sirtuina, pueden retrasar la senescencia al retirar grupos acetilo que contribuyen a la mayor accesibilidad de la cromatina.[56] La pérdida general de metilación, combinada con la adición de grupos acetilo genera una conformación de la cromatina más accesible, con una mayor tendencia a la desorganización, en comparación con las células mitóticamente activas.[48] La pérdida general de histonas impide la adición de modificaciones de histonas y contribuye al surgimiento de cambios en el enriquecimiento en ciertas regiones de la cromatina durante la senescencia.[49]
Véase también
- Epigenética
- Histona
- Nucleosoma
- cromatina
- histona acetiltransferasa
- Factor de transcripción
- CAF-1 (factor de ensamblaje de cromatina-1): chaperona de histonas que desempeña un papel de coordinación en la remodelación de la cromatina.
Referencias
- Teif VB, Rippe K (September 2009). «Predicting nucleosome positions on the DNA: combining intrinsic sequence preferences and remodeler activities». Nucleic Acids Research 37 (17): 5641-55. PMC 2761276. PMID 19625488. doi:10.1093/nar/gkp610.
- Wang GG, Allis CD, Chi P (September 2007). «Chromatin remodeling and cancer, Part I: Covalent histone modifications». Trends in Molecular Medicine 13 (9): 363-72. PMID 17822958. doi:10.1016/j.molmed.2007.07.003.
- Strahl BD, Allis CD (January 2000). «The language of covalent histone modifications». Nature 403 (6765): 41-5. Bibcode:2000Natur.403...41S. PMID 10638745. doi:10.1038/47412.
- Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ (March 2009). «Determination of enriched histone modifications in non-genic portions of the human genome». BMC Genomics 10: 143. PMC 2667539. PMID 19335899. doi:10.1186/1471-2164-10-143.
- Hublitz, Philip; Albert, Mareike; Peters, Antoine (28 de abril de 2009). «Mechanisms of Transcriptional Repression by Histone Lysine Methylation». The International Journal of Developmental Biology 10 (1387): 335-354. ISSN 1696-3547. PMID 19412890. doi:10.1387/ijdb.082717ph.
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y (September 2011). «Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification». Cell 146 (6): 1016-28. PMC 3176443. PMID 21925322. doi:10.1016/j.cell.2011.08.008.
- Jenuwein T, Allis CD (August 2001). «Translating the histone code». Science 293 (5532): 1074-80. PMID 11498575. doi:10.1126/science.1063127.
- Benevolenskaya EV (August 2007). «Histone H3K4 demethylases are essential in development and differentiation». Biochemistry and Cell Biology 85 (4): 435-43. PMID 17713579. doi:10.1139/o07-057.
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K (May 2007). «High-resolution profiling of histone methylations in the human genome». Cell 129 (4): 823-37. PMID 17512414. doi:10.1016/j.cell.2007.05.009.
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR (April 2008). «DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells». Molecular and Cellular Biology 28 (8): 2825-39. PMC 2293113. PMID 18285465. doi:10.1128/MCB.02076-07.
- Koch CM, Andrews RM, Flicek P, Dillon SC, Karaöz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I (June 2007). «The landscape of histone modifications across 1% of the human genome in five human cell lines». Genome Research 17 (6): 691-707. PMC 1891331. PMID 17567990. doi:10.1101/gr.5704207.
- Wang GG, Allis CD, Chi P (September 2007). «Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling». Trends in Molecular Medicine 13 (9): 373-80. PMC 4337864. PMID 17822959. doi:10.1016/j.molmed.2007.07.004.
- Saha A, Wittmeyer J, Cairns BR (June 2006). «Chromatin remodelling: the industrial revolution of DNA around histones». Nature Reviews Molecular Cell Biology 7 (6): 437-47. PMID 16723979. doi:10.1038/nrm1945.
- Vignali, M.; Hassan, A. H.; Neely, K. E.; Workman, J. L. (15 de marzo de 2000). «ATP-Dependent Chromatin-Remodeling Complexes». Molecular and Cellular Biology 20 (6): 1899-1910. ISSN 0270-7306. PMC 110808. PMID 10688638. doi:10.1128/mcb.20.6.1899-1910.2000.
- Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, Kozlowski M, Bultmann S, Ladurner AG, Timinszky G, Huet S (2016). «The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage». Mol. Biol. Cell 27 (24): 3791-3799. PMC 5170603. PMID 27733626. doi:10.1091/mbc.E16-05-0269.
- Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (2008). «PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites». J. Biol. Chem. 283 (2): 1197-208. PMID 18025084. doi:10.1074/jbc.M706734200.
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998). «DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139». J. Biol. Chem. 273 (10): 5858-68. PMID 9488723. doi:10.1074/jbc.273.10.5858.
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (2007). «RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins». Cell 131 (5): 887-900. PMID 18001824. doi:10.1016/j.cell.2007.09.040.
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005). «MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks». Cell 123 (7): 1213-26. PMID 16377563. doi:10.1016/j.cell.2005.09.038.
- Chapman JR, Jackson SP (2008). «Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage». EMBO Rep. 9 (8): 795-801. PMC 2442910. PMID 18583988. doi:10.1038/embor.2008.103.
- Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, Khanna KK, van Attikum H, Mailand N, Dantuma NP (2012). «A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure». EMBO J. 31 (11): 2511-27. PMC 3365417. PMID 22531782. doi:10.1038/emboj.2012.104.
- Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H (2010). «The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage». J. Cell Biol. 190 (5): 741-9. PMC 2935570. PMID 20805320. doi:10.1083/jcb.201001048.
- Hanahan D, Weinberg RA (January 2000). «The hallmarks of cancer». Cell 100 (1): 57-70. PMID 10647931. doi:10.1016/S0092-8674(00)81683-9.
- Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O (July 1998). «Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer». Nature 394 (6689): 203-6. Bibcode:1998Natur.394..203V. PMID 9671307. doi:10.1038/28212.
- Shain AH, Pollack JR (2013). «The spectrum of SWI/SNF mutations, ubiquitous in human cancers». PLOS ONE 8 (1): e55119. Bibcode:2013PLoSO...855119S. PMC 3552954. PMID 23355908. doi:10.1371/journal.pone.0055119.
- Hodges C, Kirkland JG, Crabtree GR (August 2016). «The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer». Cold Spring Harbor Perspectives in Medicine 6 (8): a026930. PMC 4968166. PMID 27413115. doi:10.1101/cshperspect.a026930.
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP (October 1994). «The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest». Cell 79 (1): 119-30. PMID 7923370. doi:10.1016/0092-8674(94)90405-7.
- Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, Sanchez-Cespedes M (May 2008). «Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines». Human Mutation 29 (5): 617-22. PMID 18386774. doi:10.1002/humu.20730.
- Hodges HC, Stanton BZ, Cermakova K, Chang CY, Miller EL, Kirkland JG, Ku WL, Veverka V, Zhao K, Crabtree GR (January 2018). «Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers». Nature Structural & Molecular Biology 25 (1): 61-72. PMC 5909405. PMID 29323272. doi:10.1038/s41594-017-0007-3.
- Stanton BZ, Hodges C, Calarco JP, Braun SM, Ku WL, Kadoch C, Zhao K, Crabtree GR (February 2017). «Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin». Nature Genetics 49 (2): 282-288. PMC 5373480. PMID 27941795. doi:10.1038/ng.3735.
- Liquori, Alessandro; Ibañez, Mariam; Sargas, Claudia; Sanz, Miguel Ángel; Barragán, Eva; Cervera, José (8 de marzo de 2020). «Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene». Cancers 12 (3): 624. ISSN 2072-6694. PMC 7139833. PMID 32182684. doi:10.3390/cancers12030624.
- Wolffe AP (May 2001). «Chromatin remodeling: why it is important in cancer». Oncogene 20 (24): 2988-90. PMID 11420713. doi:10.1038/sj.onc.1204322.
- Tu, William B.; Helander, Sara; Pilstål, Robert; Hickman, K. Ashley; Lourenco, Corey; Jurisica, Igor; Raught, Brian; Wallner, Björn et al. (May 2015). «Myc and its interactors take shape». Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1849 (5): 469-483. ISSN 0006-3002. PMID 24933113. doi:10.1016/j.bbagrm.2014.06.002.
- Marks PA, Dokmanovic M (December 2005). «Histone deacetylase inhibitors: discovery and development as anticancer agents». Expert Opinion on Investigational Drugs 14 (12): 1497-511. PMID 16307490. doi:10.1517/13543784.14.12.1497.
- Richon VM, O'Brien JP (2002). «Histone deacetylase inhibitors: a new class of potential therapeutic agents for cancer treatment». Clinical Cancer Research 8 (3): 662-4. PMID 11895892.
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA (August 2000). «Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation». Proceedings of the National Academy of Sciences of the United States of America 97 (18): 10014-9. Bibcode:2000PNAS...9710014R. PMC 27656. PMID 10954755. doi:10.1073/pnas.180316197.
- Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S, Iwase H (November 2005). «Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*». Breast Cancer Research and Treatment 94 (1): 11-6. PMID 16172792. doi:10.1007/s10549-005-6001-1.
- Munshi, Anupama; Tanaka, Toshimitsu; Hobbs, Marvette L.; Tucker, Susan L.; Richon, Victoria M.; Meyn, Raymond E. (August 2006). «Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci». Molecular Cancer Therapeutics 5 (8): 1967-1974. ISSN 1535-7163. PMID 16928817. doi:10.1158/1535-7163.MCT-06-0022.
- «Zolinza® (vorinostat) Capsules. Full Prescribing Information». Food and Drug Administration (FDA). Archivado desde el original el 9 de febrero de 2023. Consultado el 9 de febrero de 2023.
- VanderMolen, Karen M.; McCulloch, William; Pearce, Cedric J.; Oberlies, Nicholas H. (August 2011). «Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): a natural product recently approved for cutaneous T-cell lymphoma». The Journal of Antibiotics 64 (8): 525-531. ISSN 1881-1469. PMC 3163831. PMID 21587264. doi:10.1038/ja.2011.35.
- «ISTODAX® (romidepsin) for injection, for intravenous use. Full prescribing information». Food and Drug Administration (FDA). Archivado desde el original el 9 de febrero de 2023. Consultado el 9 de febrero de 2023.
- Dowden J, Hong W, Parry RV, Pike RA, Ward SG (April 2010). «Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases». Bioorganic & Medicinal Chemistry Letters 20 (7): 2103-5. PMID 20219369. doi:10.1016/j.bmcl.2010.02.069.
- Wong, Lee H.; McGhie, James D.; Sim, Marcus; Anderson, Melissa A.; Ahn, Soyeon; Hannan, Ross D.; George, Amee J.; Morgan, Kylie A. et al. (March 2010). «ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells». Genome Research 20 (3): 351-360. ISSN 1549-5469. PMC 2840985. PMID 20110566. doi:10.1101/gr.101477.109.
- Clapier CR, Cairns BR (2009). «The biology of chromatin remodeling complexes». Annual Review of Biochemistry 78: 273-304. PMID 19355820. doi:10.1146/annurev.biochem.77.062706.153223.
- Parry, Aled John; Narita, Masashi (2016). «Old cells, new tricks: chromatin structure in senescence». Mammalian Genome 27 (7–8): 320-331. ISSN 0938-8990. PMC 4935760. PMID 27021489. doi:10.1007/s00335-016-9628-9.
- Hayflick, L.; Moorhead, P. S. (1 de diciembre de 1961). «The serial cultivation of human diploid cell strains». Experimental Cell Research 25 (3): 585-621. ISSN 0014-4827. PMID 13905658. doi:10.1016/0014-4827(61)90192-6.
- Chandra, Tamir; Ewels, Philip Andrew; Schoenfelder, Stefan; Furlan-Magaril, Mayra; Wingett, Steven William; Kirschner, Kristina; Thuret, Jean-Yves; Andrews, Simon et al. (29 de enero de 2015). «Global Reorganization of the Nuclear Landscape in Senescent Cells». Cell Reports 10 (4): 471-483. ISSN 2211-1247. PMC 4542308. PMID 25640177. doi:10.1016/j.celrep.2014.12.055.
- Sun, Luyang; Yu, Ruofan; Dang, Weiwei (16 de abril de 2018). «Chromatin Architectural Changes during Cellular Senescence and Aging». Genes 9 (4): 211. ISSN 2073-4425. PMC 5924553. PMID 29659513. doi:10.3390/genes9040211.
- Criscione, Steven W.; Teo, Yee Voan; Neretti, Nicola (2016). «The chromatin landscape of cellular senescence». Trends in Genetics 32 (11): 751-761. ISSN 0168-9525. PMC 5235059. PMID 27692431. doi:10.1016/j.tig.2016.09.005.
- Yang, Na; Sen, Payel (3 de noviembre de 2018). «The senescent cell epigenome». Aging (Albany NY) 10 (11): 3590-3609. ISSN 1945-4589. PMC 6286853. PMID 30391936. doi:10.18632/aging.101617.
- Basta, Jeannine; Rauchman, Michael (2015). «The Nucleosome Remodeling and Deacetylase (NuRD) Complex in Development and Disease». Translational Research 165 (1): 36-47. ISSN 1931-5244. PMC 4793962. PMID 24880148. doi:10.1016/j.trsl.2014.05.003.
- Li, Xueping; Ding, Dong; Yao, Jun; Zhou, Bin; Shen, Ting; Qi, Yun; Ni, Ting; Wei, Gang (15 de julio de 2019). «Chromatin remodeling factor BAZ1A regulates cellular senescence in both cancer and normal cells». Life Sciences 229: 225-232. ISSN 1879-0631. PMID 31085244. doi:10.1016/j.lfs.2019.05.023.
- López-Otín, Carlos; Blasco, Maria A.; Partridge, Linda; Serrano, Manuel; Kroemer, Guido (6 de junio de 2013). «The Hallmarks of Aging». Cell 153 (6): 1194-1217. ISSN 0092-8674. PMC 3836174. PMID 23746838. doi:10.1016/j.cell.2013.05.039.
- Tasdemir, Nilgun; Banito, Ana; Roe, Jae-Seok; Alonso-Curbelo, Direna; Camiolo, Matthew; Tschaharganeh, Darjus F.; Huang, Chun-Hao; Aksoy, Ozlem et al. (2016). «BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance». Cancer Discovery 6 (6): 612-629. ISSN 2159-8290. PMC 4893996. PMID 27099234. doi:10.1158/2159-8290.CD-16-0217.
- Wilson, V. L.; Jones, P. A. (3 de junio de 1983). «DNA methylation decreases in aging but not in immortal cells». Science 220 (4601): 1055-1057. Bibcode:1983Sci...220.1055W. ISSN 0036-8075. PMID 6844925. doi:10.1126/science.6844925.
- Kaeberlein, Matt; McVey, Mitch; Guarente, Leonard (1 de octubre de 1999). «The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms». Genes & Development 13 (19): 2570-2580. ISSN 0890-9369. PMC 317077. PMID 10521401. doi:10.1101/gad.13.19.2570.
Otras lecturas
- Chen T, Dent SY (February 2014). «Chromatin modifiers and remodellers: regulators of cellular differentiation». Nature Reviews Genetics 15 (2): 93-106. PMC 3999985. PMID 24366184. doi:10.1038/nrg3607.
Enlaces externos
- MBInfo - Cromatina
- MBInfo - Envasado de ADN
- YouTube - Cromatina, Histonas y Modificaciones
- YouTube: descripción general de la epigenética
- MeSH: Chromatin+remodeling (en inglés)
| Control de autoridades |
|
|---|
 Datos: Q5113739
Datos: Q5113739