Acides aminés D
Les acides aminés D sont une classe d'acides aminés, ou pour ce qui intéresse le plus la biologie, les acides α-aminés, pour lesquels les groupes fonctionnels carboxyle (-COOH) et amine (-NH2) sont liés à un carbone α en configuration D par rapport d'une part à une chaîne latérale dépendant du type d'acide et d'autre part à un atome d'hydrogène. Ce sont les énantiomères des acides aminés L.
Dans tous les systèmes biologiques, les acides aminés D sont bien plus rares que leurs isomères L, qui sont les briques de construction importantes de la matière vivante, sous la forme des 22 acides aminés protéinogènes. C'est pourquoi on en a longtemps déduit que les acides aminés D n'ont pas de fonction biologique, et ne sont pas « naturels ». Ce tableau s'est complètement modifié à partir du début des années 1990. Aujourd'hui, on sait que des acides aminés D sont contenus par exemple dans des antibiotiques peptidiques sécrétés par des bactéries, ou bien diverses plantes, telles le riz, l'ail ou les pois.
Certains acides aminés D remplissent aussi d'importantes fonctions physiologiques chez l'homme. En particulier, dans le système nerveux central, ce sont la D-sérine et le D-aspartate. Mais les acides aminés D semblent aussi jouer un rôle dans certaines maladies, comme dans la schizophrénie. Ce domaine de recherche est relativement nouveau, et beaucoup de fonctions des acides aminés D libres ou liés dans des peptides ou des protéines sont encore largement inconnues, ou incomprises.
Au moyen de procédés d'analyse chromatographiques, on a pu mettre en évidence des acides aminés D dans une série d'aliments et d'organismes. Une application en est la datation par racémisation des acides aminés pour la détermination de l'âge des fossiles.
Selon l'état actuel[Quand ?] des recherches, les acides aminés D libres absorbés journellement avec la nourriture, ne sont pas dangereux pour l'homme. Beaucoup de médicaments importants contiennent des acides aminés D. Des acides aminés D produits artificiellement sont utilisés comme éléments pour la fabrication d'antibiotiques (semi-)synthétiques et d'un grand nombre de produits de la vie courante, et notamment de médicaments.
Fondements
Chiralité
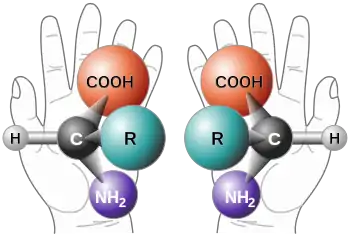
Tous les acides aminés protéinogènes (spécifiés par le code génétique), à l'exception de la glycine, le plus simple d'entre eux, possèdent un atome de carbone, lié à quatre radicaux différents. Ces radicaux occupent dans l'espace les quatre sommets d'un tétraèdre. Cet arrangement cause une asymétrie, ce qui a pour conséquence deux possibilités pour leur arrangement relatif. Ces deux formes, appelées énantiomères ou isomères par réflexion, se comportent comme un objet et son image dans le miroir. L'atome de carbone asymétrique y forme le centre asymétrique. L'énantiomère et son image miroir ne sont pas superposables. Ceci arrive aussi pour des objets de la vie courante qui n'ont pas de plan de symétrie. Un exemple en est les deux mains. Les mains droite et gauche sont comme un objet et son image dans le miroir, mais elles ne sont pas superposables. Leur différence devient particulièrement importante quand ils interagissent avec d'autres systèmes chiraux (du mot grec pour main). Par exemple quand une main droite tente de prendre une autre main droite ou gauche, ou essaie d'enfiler un mauvais gant. Dans des environnements chiraux, les différences entre énantiomères moléculaires apparaissent particulièrement clairement.
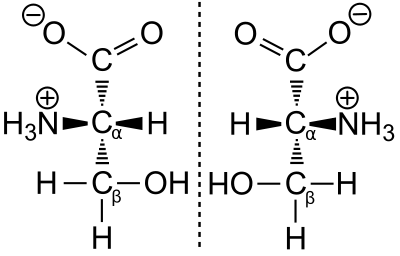
Le Prix Nobel de chimie Emil Fischer a mis au point un procédé de projection, la projection de Fischer, par laquelle on peut décrire de façon univoque en deux dimensions la structure spatiale d'une liaison chimique chirale. Pour cela, il a choisi une substance de référence (le glycéraldéhyde). Selon les règles de la projection de Fischer, le groupe acide (carboxyle) est toujours représenté en haut, et le radical R qui fait la différence entre acides aminés toujours en bas. Si le groupe amine se situe dans cette projection à gauche (en latin laevus), on parle d'un acide aminé L. Si le groupe amine est à droite (latin : dexter) en projection de Fischer, il s'agit d'un acide aminé D. Les adjectifs gauche et droit ne se rapportent qu'à la configuration en projection de Fischer.
Dans leurs propriétés physiques, comme le point de fusion, la masse spécifique, la solubilité dans l'eau ou autres milieux, le pH isoélectrique, les acides aminés D et L sont totalement identiques. De plus, dans un milieu achiral, c'est-à-dire dans un environnement dépourvu d'autres molécules chirales, ils se comportent exactement de la même manière, à une exception près : à conditions identiques, deux énantiomères font tourner le plan de polarisation de la lumière polarisée linéairement d'une quantité égale en valeur absolue, mais en sens opposé. S'ils la font tourner dans le sens des aiguilles d'une montre (pour un observateur faisant face à la source de lumière), on parle de forme dextrogyre ou forme (+). Dans le cas opposé, il s'agit de forme lévogyre, ou (-). Ces distinctions ne jouent pratiquement aucun rôle dans la vie courante. Dans la littérature, des confusions arrivent même entre formes L et formes (-). De plus, la valeur du pouvoir rotatoire, et même son signe, dépendent fortement des conditions. Par exemple, l'acide aminé L-leucine à température ambiante en solution de molarité 6 M d'acide chlorhydrique a un pouvoir rotatoire spécifique de +15,1° (à droite), et dans l'eau pure de -10,8° (à gauche). En solution de soude de molarité 3 M, elle est à nouveau à +7,6° (à droite)[1].
Un mélange de 50 % d'énantiomère D et de 50 % de L s'appelle un racémique. Les racémiques sont notamment fabriqués dans les opérations de synthèse artificielle d'acides aminés. Ils n'ont pas d'activité optique, c'est-à-dire qu'ils ne changent pas le plan de polarisation de la lumière. Les racémiques ont en partie des propriétés physiques différentes des énantiomères purs (par exemple, le point de fusion), mais couramment des propriétés physiologiques différentes.
Conventions de dénomination et nomenclature
La projection de Fischer est jusqu'à présent le système préféré de projection pour les acides aminés et les glucides. À côté, il existe pour les acides aminés la convention de Cahn-Ingold-Pregold (Système CIP), qui décrit la configuration de molécules chirales. Dans ce système, la plupart des acides aminés L protéinogènes sont des acides aminés S. Leurs énantiomères, les acides aminés D sont pour la plupart en configuration R. Les exceptions sont la L-cystéine, la L-cystine et la L-sélénocystéine, parce que le soufre ou le sélénium a dans la nomenclature CIP une priorité plus élevée que l'oxygène. Ces trois acides aminés L sont dans une configuration R. Leurs énantiomères ont par contre une configuration S.
Dans les séquences d'acides aminés, les acides aminés D sont notés dans l'abréviation à 3 lettres par un préfixe D en petite capitale.
Par exemple, l'heptapeptide dermorphine :
H-Tyr-D-Ala-Phe-Gly-Tyr-Pro-Ser-NH2.
Dans les codes canoniques à une lettre, les acides aminés D sont représentés par la minuscule de la lettre correspondant à l'acide aminé L correspondant.
Par exemple, la dermorphine :
YaFGYPS-NH2.
Présence naturelle et histoire de la découverte
Les acides aminés D sont bien plus rares dans la nature que leurs énantiomères L, qui forment, avec les acides nucléiques, les pierres de construction de la vie. Une asymétrie du même genre, avec l'apparition de deux énantiomères, se présente pour les glucides. Dans ce dernier cas, c'est la forme D, par exemple le D-glucose, qui est la configuration « naturelle ». On estime la quantité de D-glucose sur Terre égale à 1015 fois celle de L-glucose[2]. Pour les acides aminés, il n'y a pas encore d'estimation fiable.
Pendant longtemps, on est parti du fait que seuls les acides aminés L avaient été sélectionnés au cours de l'évolution pour la formation de peptides et de protéines[3]. Depuis les années 1980, des méthodes d'analyse améliorées ont conduit à réviser cette hypothèse. Des acides aminés D ont été trouvés dans toujours plus d'êtres vivants, si bien qu'ils ont une diffusion et une variété bien plus grande que ce qui avait été supposé tout d'abord. Dans la littérature récente, les acides aminés D sont maintenant considérés comme composants ordinaires de plantes et d'aliments[2]. Et même chez des êtres vivants supérieurs, jusque chez l'homme, les acides aminés D sont impliqués dans des processus physiologiques importants, qui sont encore largement incompris[4].
Le développement de la vie sur Terre supposait une homochiralité, c'est-à-dire une configuration homogène des acides aminés et des autres pierres de construction de la vie. Dans un milieu racémique, il ne peut y avoir aucune réplication[2],[5],[6]. Il y a toute une série d'hypothèses sur la cause du déséquilibre extrême entre les quantités des deux formes énantiomères des acides aminés. Il y a unanimité sur le fait qu'il y aurait eu dans la nature un premier déséquilibre entre acides aminés D et L. À partir de là, on peut très bien expliquer l'extrême enrichissement de l'une des deux formes, par amplification chirale, c'est-à-dire un effet d'auto-amplification qui conduit dans une réaction chimique, en présence d'un léger excès d'une des formes énantiomères, à un résultat encore plus déséquilibré. Il reste néanmoins totalement à résoudre le problème du fait qu'une symétrie initiale aurait été brisée, très probablement bien avant le début de la vie sur Terre. On a discuté, parmi les sources possibles, de la brisure de la symétrie de parité dans la radioactivité β (hypothèse de Vester-Ulbricht)[7],[8] ou la contamination de la soupe primordiale par des excès d'acides aminés L extraterrestres.
En faveur de cette dernière théorie, parle le fait que dans la météorite de Murchison, on a mis en évidence un excès d'acides aminés L non protéinogènes : acide 2-Amino-2,3-diméthylpentanoïque[9] et isovaline[10],[11]. Dans la météorite de Murchison, l'excès en L-isovaline était d'environ 18,5 %, et dans celle d'Orgueil, de 15,2 %[12]. Ces excès ont pu être provoqués par un rayonnement UV polarisé circulairement, qui — comme on le confirme expérimentalement — détruit préférentiellement les acides aminés D[13].
Formation d'acides aminés D par racémisation
De grandes quantités d'acides aminés D peuvent provenir d'acides aminés L par racémisation. La formation d'un racémique d'acides aminés, c'est-à-dire d'un mélange à parties égales d'acides aminés D et L, est privilégiée thermodynamiquement. Certes, l'enthalpie reste inchangée, mais le degré supérieur de « désordre » conduit à une augmentation de l'entropie, ce qui amène une diminution de l'enthalpie libre G de ΔG. La valeur de cette décroissance est de -1,6 kJ/mole à 25 °C[14].
Des températures supérieures conduisent à une plus grande perte d'enthalpie libre, ce pourquoi la racémisation est substantiellement accélérée. La demi-vie de la racémisation, définie comme le temps dans lequel la valeur de l'excès d'énantiomère diminue de moitié, dépend, à côté de la température, du pH, du type d'acide aminé, du milieu solvant, de l'humidité et de la présence de catalyseurs. Dans des conditions constantes, la racémisation peut bien se calculer, et inversement, du degré de racémisation, on peut tirer des conclusions sur l'âge de l'échantillon. Ce procédé, désigné par « datation par racémisation des acides aminés » peut être utilisé pour la datation de fossiles, mais aussi pour des organismes vivants. À la mort, tous les processus qui luttent contre la racémisation des acides aminés dans l'organisme concerné s'arrêtent. La vie est une lutte contre l'entropie[15], et les processus qui vont à l'encontre de la racémisation s'arrêtent au plus tard à la mort. Dans certains tissus, où le métabolisme des protéines est très faible, ce processus commence déjà avant la fin de la formation du tissu. Un exemple en est le collagène de la dentine des dents, ou le cristallin de l'œil[16]. La température et le pH relativement constants dans les dents permettent de définir l'âge d'un organisme vivant par le degré de racémisation de l'aspartate avec une précision de ±4 ans environ[17],[18]. Ce procédé est notamment utilisé en médecine légale[19]. Un exemple pour la performance de cette méthode est la recherche faite en 1996 sur les ossements de l'empereur Lothaire de Supplinbourg (1075–1137)[20]. Dans cette étude, on a trouvé chez Lothaire un degré de racémisation notablement plus élevé que pour son épouse Richenza von Northeim et de son gendre Henri X de Bavière, ce qui correspondrait à un âge d'environ 9000 ans de plus. Le taux de racémisation des deux témoins correspondait par contre très bien à leur âge d'environ 850 ans. Dans les trois cas, c'est le taux de racémisation de l'aspartate qui a été mesuré. Le degré élevé de racémisation de Lothaire est à rapporter aux circonstances particulières de sa mort. Il est mort près de Breitenwang dans le Tyrol, à peu près à 700 km de son siège à Königslutter am Elm. Pour protéger son corps de la pourriture pour ce long trajet, le cadavre a été traité selon le Mos Teutonicus (« usage teuton »). Ceci consiste à cuire le cadavre, à enlever la chair des os, et à ne transporter que les ossements. La cuisson a racémisé le L-aspartate, mesuré 859 ans plus tard, notablement plus fort que pour les cadavres normalement enterrés de sa femme et de son gendre. Par le degré de racémisation, on a pu estimer la durée de cuisson à environ six heures[2].
Dans les cheveux du cadavre âgé d'environ 5300 ans de l'homme des Alpes de l'Ötztal, mieux connu sous le surnom d'« Ötzi », 37 % de l'hydroxyproline apparaissent en configuration D. Dans une momie vieille de 3000 ans, c'était 31 %, dans des cheveux du Moyen Âge (environ 1000 ans) 19 % et dans des échantillons modernes de 4 %[21].
Pendant la préparation de la nourriture, les acides aminés L peuvent aussi être racémisés dans les protéines, en raison de la température et de valeurs extrêmes du pH. Les acides aminés différents se racémisent différemment vite. La vitesse de racémisation dépend fortement de la chaîne latérale de l'acide aminé et des acides aminés voisins. Les groupes qui attirent les électrons facilitent la protonation de l'atome C α, ce qui facilite la racémisation[22],[23]. Ceci est notamment pertinent pour la sérine et l'aspartate. En outre, des effets stériques jouent un rôle[24]. L'asparagine et l'aspartate se racémisent particulièrement vite quand ils sont au voisinage d'une glycine dans la séquence peptidique. Il peut alors se produire un succinimide cyclique, ce qui, du point de vue thermodynamique, favorise fortement la transformation d'un énantiomère dans l'autre[25],[26]. Pour des basses valeurs du pH, par exemple dans une solution 6M d'acide chlorhydrique, l'aspartate se racémise le plus vite. La proline et la glutamine se racémisent notablement plus lentement, tandis que dans ces conditions, l'isoleucine, la valine, la sérine et la thréonine ne se racémisent que très peu. Dans une solution de soude 1M, la sérine se racémise le plus vite, puis l'aspartate, la phénylalanine, le glutamate et la valine[26],[27].
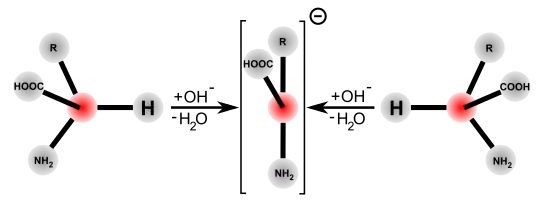
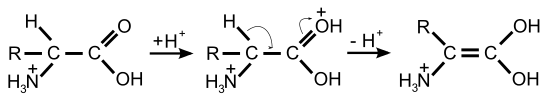
La racémisation catalysée par les bases ou les acides nécessite des conditions très drastiques pour obtenir une racémisation complète en quelques heures. Par opposition, la racémisation catalysée par des enzymes dans les systèmes biologiques est notablement plus rapide et se déroule dans des conditions plus douces : pH à peu près neutre, et température ambiante ou corporelle. Les racémases catalysent la déprotonation du carbone α de l'acide aminé. Dans cette position, l'atome d'hydrogène n'est plus que très faiblement acide. La constante d'acidité de la forme protonée a un pKs ≈ 21, et est au point isoélectrique encore plus faible avec ≈ 29[29],[30]. La déprotonation est facilitée pour la plupart des racémases par le phosphate de pyridoxal (PALP). Sur le centre actif de ces enzymes, le PALP est lié à un résidu de lysine. Le groupe amino de l'acide aminé L se lie alors au groupe aldéhyde du PALP et forme alors une imine (aldimine, ou base de Schiff). Comme catalyseur électrophile, le PALP tire sur le cycle aromatique des électrons extraits du carbone α de l'acide aminé, qui est alors plus facile à déprotoner. En outre, l'anion restant est stabilisé par effet mésomère. Une reprotonation avec addition d'eau libère alors l'acide aminé racémisé comme produit de la réaction par hydrolyse de la base de Schiff[31].
Outre cela, il y a aussi des racémases indépendantes du PALP, dans le centre actif desquelles les groupes thiol de deux cystéines catalysent la protonation[32],[33]. Dans un mécanisme à deux bases, un thiolate déprotoné (R-S−) jouant le rôle de base, capte le proton du carbone α. Le groupe thiol de l'autre cystéine est chargé de la reprotonation[31]. Ces processus de racémisation catalysés par des enzymes produisent la plus grande partie des acides aminés D dans les organismes.

Antibiotiques peptidiques et autres médicaments peptidiques d'origine naturelle
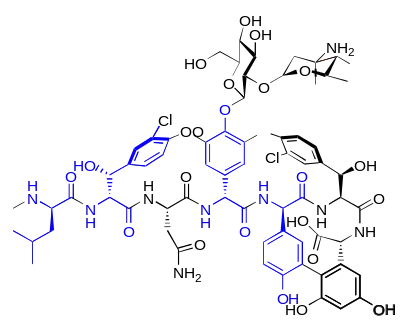
Un grand nombre d'antibiotiques peptidiques sont construits à partir d'acides aminés D. Ce sont des substances naturelles, fabriquées par des procaryotes au moyen de synthèses non-ribosomiques[31]. Le groupe très important du point de vue pharmacologique des pénicillines contient comme élément de structure élémentaire la D-pénicillamine, un acide aminé non protéinogène. Les polymyxines et les actinomycines sont aussi construites autour d'acides aminés D (respectivement D-phénylalanine et D-valine). La bacitracine produite par le Bacillus subtilis consiste notamment d'aspartate, de glutamate, d'ornithine et de phénylalanine en configuration D. La valinomycine produite par Streptomyces fulvissimus contient de la D-valine, et la circuline A formée par le bacillus circulans contient de la D-leucine. Parmi les antibiotiques à acides aminés D, on compte encore notamment : la fongisporine (D-phénylalanine et D-valine), la gramicidine et la tyrocidine (D-phénylalanine), la malformine C (D-leucine et D-cystéine ), la mycobacilline (D-aspartate et D-glutamate)[34].
Des champignons ascomycètes produisent aussi des médicaments naturels contenant des acides aminés D, comme le tolypocladium inflatum, produisant l'immunosuppresseur ciclosporine avec de la D-alanine, ou le penicillium chrysogenum, produisant la pénicilline M avec de la D-valine.
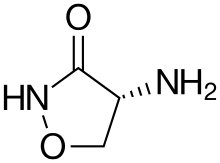
La cyclosérine utilisée dans le traitement de la tuberculose, relativement simple chimiquement, est produite par des streptomycètes, comme Streptomyces garyphalus, avec de la D-sérine.
Acides aminés D et peptides contenant des acides aminés D

Pendant longtemps, on est parti de l'hypothèse que, dans la nature, domine un seul énantiomère des acides aminés, c'est-à-dire la forme L. Jusqu'aux années 1960, les acides aminés D étaient considérés comme des artéfacts de laboratoire (erreurs dépendant du système), et rangés parmi les « isomères non-naturels ». Aujourd'hui encore on trouve cette désignation d'« isomères non-naturels » pour les énantiomères D[35].
Oxydases des acides aminés D – enzymes sans substrat ?
En 1933, le médecin et chimiste allemand, futur Prix Nobel de physiologie ou médecine, Hans Adolf Krebs découvre l'enzyme oxydase des acides aminés D[36], et la décrit en détail deux ans plus tard[37],[38]. Krebs établit que les acides aminés D, qui « n'apparaissent pas dans la nature » sont désaminés nettement plus vite que leurs isomères « naturels » L en présence de rein ou de foie de porc frais pressé. Avec un inhibiteur choisi, par exemple avec de l'octan-1-ol, il pouvait désactiver l'oxydase d'acide aminé L, et aboutir ainsi à ne plus désaminer sélectivement que les acides aminés D. Krebs en conclut que dans les organes utilisés, ou dans leurs extraits, il se trouvaient deux oxydases d'acides aminés, qui agissaient respectivement sur les acides aminés L et sur les D. Krebs se montra étonné qu'il y ait une enzyme qui n'agisse exclusivement que sur des substances non naturelles. Mais il fit remarquer que Félix Ehrlich en 1914[39], Edmund von Lippmann en 1884[40] et Sigmund Fränkel en 1923/24[41] avaient signalé l'apparition occasionnelle d'acides aminés D dans la nature[37]. La D-alanine isolée du cèpe de Bordeaux par E. Winterstein et al en 1913[42], a été une de ces preuves précoces[43].
Acides aminés D dans les plantes
Dans les plantes, on peut montrer l'existence d'acides aminés D tant libres que liés dans des chaînes peptidiques[44]. Ils sont souvent contenus dans les plantes sous forme de dérivés N-malonyle ou N-acétyle.
Par exemple, dans la racide de tournesol (Helianthus annuus) 40 % de l'alanine est en configuration D. La D-alanine et le dipeptide D-Ala-D-Ala se trouvent dans diverses herbes[45] ; de même dans le riz (Oryza australiensis), où environ 10 % de la sérine est dans la conformation D. Elle est fabriquée par la plante elle-même avec une racémase de la sérine. Le gène correspondant pour cette enzyme, chez l'Oryza sativa ssp. Japonica cv. Nipponbare, se situe sur le chromosome 4[46]. On a montré des acides aminés D en plus faibles concentrations dans une multitude de plantes alimentaires. On y compte notamment les pois (Pisum sativum)[47], l'ail, diverses espèces de chou et de fruits[48],[49]. Il n'est encore pas du tout clair de savoir quelle fonction ces acides aminés D libres et peptidiques jouent dans les plantes[26].

Bactéries et acides aminés D
Avant la découverte d'acides aminés D libres, on a identifié des acides aminés D dans une série de composés d'origine microbienne. Par exemple, la pénicilline G, formée dans des cultures de moisissures penicillium notatum, découverte en 1928 par Alexander Fleming, contient comme élément notable la D-pénicillamine (ou 3-mercapto-D-valine).
Le Texan Esmond E. Snell a remarqué en 1943 dans des expériences sur les cultures d'enterococcus faecalis et de lactobacillus casei que pour la croissance de ces espèces de bactéries, on peut remplacer complètement la pyridoxine (vitamine B6) nécessaire par de la D-alanine dans l'alimentation[50],[51],[52]. Il établit en outre que la D-alanine est notablement plus efficace dans ce rôle que la L-alanine[53]. Quand on a pu démontrer ensuite que de grandes quantités de D-alanine étaient présentes dans les peptidoglycanes - les biopolymères qui donnent à la paroi cellulaire des bactéries leur solidité - il a été clair pourquoi les cellules avaient besoin de ces acides aminés « non-naturels. » L'inclusion de D-alanine, et surtout de D-glutamate, empêche la destruction des peptidoglycanes par les peptidases. De façon intéressante, c'est précisément cette « digue de protection » d'acides aminés D qui forme le point d'attaque pour les antibiotiques β-lactame, comme la pénicilline. Ces antibiotiques inhibent l'enzyme transpeptidase de la D-alanine, qui se trouve exclusivement dans les bactéries, et qui catalyse la réticulation des peptidoglycanes, spécialement par la D-alanine[54].
En 1951, Irwin Clyde Gunsalus et Willis A. Wood ont isolé de l'enterococcus faecalis une racémase de l'alanine, enzyme qui catalyse la racémisation de la L-alanine naturelle[55]. Le gène alr qui code la racémase de l'alanine est présent chez toutes les bactéries[56]. La D-alanine formée à l'aide de la racémase de l'alanine est essentielle pour la synthèse des peptidoglycanes chez pratiquement toutes les bactéries[57]. Outre la D-alanine et le D-glutamate, on trouve aussi chez certaines espèces d'entérocoques de la D-sérine dans la paroi cellulaire[58],[59]. Cette D-sérine y forme avec la D-alanine à l'extrémité C-terminale un dipeptide D-Ala-D-Ser, qui est responsable de la résistance de ces espèces de bactéries aux glycopeptides, comme la vancomycine[60],[61].
Acides aminés D dans les éponges
On a pu trouver dans les éponges des polythéonamides. Ce sont des toxines peptidiques dont les acides aminés alternent entre formes D et L. Ils sont apparemment synthétisés par les ribosomes sous forme de peptides L, puis, après la traduction, un acide aminé sur deux est épimérisé. Ceci se produit au moyen de quelques enzymes, dont les gènes, provenant apparemment de bactéries, sont arrivés chez les éponges par transfert horizontal de gènes[62],[63].
Acides aminés D chez les organismes multicellulaires
Dankwart Ackermann et M. Mohr ont pu trouver en 1937 dans le foie du requin épineux de la D-ornithine[64]. L'oxydase des acides aminés D découverte par Krebs a été découverte dans les années suivantes chez tous les mammifères. H. Blaschko et Joyce Hawkins l'ont trouvée pour la première fois en 1951 chez des invertébrés. Mais la fonction de cet enzyme dans les divers organismes est restée encore non clarifiée[38]. Vers la fin des années 1960, on a spéculé que l'enzyme servait dans le tube digestif à la destruction des composants des parois cellulaires des bactéries gram-positives, qui contiennent de grandes quantités d'acides aminés D[65]. La théorie que l'oxydase des acides aminés D ne servait qu'à la destruction d'acides aminés apportés de l'extérieur (exogènes) a persisté jusqu'au début des années 1990.
C'est en 1950 qu'Auclair et Patton[66] ont trouvé pour la première fois de la D-alanine chez un organisme multicellulaire, dans l'hémolymphe de la punaise Oncopeltus fasciatus. Ils ont utilisé pour faire l'analyse une chromatographie sur papier à deux dimensions. Après l'élution, ils ont pulvérisé les chromatogrammes séchés avec de l'oxydase des acides aminés D, qui ne désamine que la D-alanine en acide cétocarbonique, que l'on identifie facilement avec de la phénylhydrazine[52]. On supposa la présence de D-alanine due à la flore microbienne, à une contamination par la nourriture, ainsi qu'une racémisation spontanée due à l'âge[67].
La biosynthèse de D-sérine a été démontrée en 1965 par un groupe de travail autour de John J. Corrigan à l'Université Tufts au Massachusetts. Les vers à soie alimentésavec du D-glucose marqué radioactivement produisent aussi bien de la D-sérine que de la L-sérine[68]. Plus tard, on a trouvé des acides aminés D dans d'autres insectes[69] et mammifères[70].

En 1962, un groupe italien autour de Vittorio Erspamer a isolé dans la
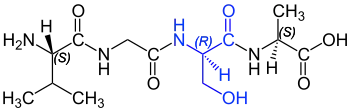
l'anoure sud-américain Physalaemus fuscomaculatus la tachykinine physalémine[72]. Ce polypeptide est composé de douze acides aminés, et commence de l'extrémité N-terminale, par une D-proline. En code à une lettre, la séquence s'écrit pEADPNKFYGLM-NH2[73]. C'est le premier peptide naturel découvert avec un acide aminé D qui ne soit pas d'origine microbienne. Mais même, trois ans après, le biochimiste américain Alton Meister écrivait par exemple dans son ouvrage standard Biochemistry of the amino acids, « qu'il n'y a actuellement aucune preuve décisive de l'existence d'acide aminé D dans les protéines des plantes et des animaux[74],[21]. » On n'a tout d'abord fait à peine attention à la découverte d'Erspamer. Ce n'est que quand le même groupe a, 19 ans plus tard, isolé la dermorphine chez la Phyllomedusa sauvagii, aussi originaire d'Amérique du Sud[75],[76], que l'on a commencé lentement à reconnaître la portée de cette découverte. À partir de l'extrémité N-terminale, la dermorphine, constituée de sept acides aminés contient en position 2 une D-alanine. La configuration D de cette alanine est indispensable pour l'activité pharmacologique. La dermorphine se lie au récepteur µ1 et y est nettement plus sélective et puissante que les endorphines endogènes (dynorphine et enképhaline) et que la morphine très répandue, d'origine végétale[77]. La découverte contredisait un certain nombre de paradigmes, si bien qu'Erspamer a eu des difficultés certaines à trouver un journal spécialisé qui accepte de publier ses travaux[78],[79]. Un de ces paradigmes est que, dans la biosynthèse des protéines, l'ADN d'un organisme ne code que les 22 acides aminés protéinogènes, qui sont tous en conformation L. Il n'y a pas de gène pour coder des acides aminés D. Ce n'est que plus de dix ans plus tard que la contradiction fut résolue : c'est une modification post-traductionnelle stéréosélective catalysée par des épimérases qui est responsable de l'apparition d'acides aminés D dans des peptides d'eucaryotes. C'est-à-dire qu'après la traduction, la conformation d'un acide aminé L déterminé est modifiée par une enzyme spécifique endogène[80].
Acides aminés D chez les mammifères
Jusqu'en 1992, il était exclu que les acides aminés D aient une fonction biologique chez lez mammifères[67]. Par amélioration des procédés de mesure analytique comme la chromatographie en phase gazeuse[81] ou liquide à haute performance[82],[49], il est devenu possible à partir des années 1980 de séparer proprement les acides aminés D de leurs énantiomères L, et de les mettre encore en évidence en très petite quantité. Atsushi Hashimoto et al ont trouvé ainsi en 1992 dans le cerveau de rats domestiques de relativement grandes quantités de D-sérine libre. Ils indiquèrent une concentration d'environ 0,27 µmol/g de masse cérébrale, ce qui impliquait un rapport de D-sérine/L-sérine de 0,23[83]. Il était déjà connu auparavant que de la D-sérine apportée de l'extérieur (exogène) était un agoniste allostérique puissant et sélectif du récepteur NMDA (NMDA = N-méthyl-D-aspartate)[84]. La source de concentration relativement élevée en D-sérine, qui a été démontrée par la suite aussi dans le cerveau d'autres mammifères, y compris l'homme, est tout d'abord restée incertaine. Des spéculations, comme l'absorption ciblée de L-sérine racémisée dans la nourriture, et son transport vers le cerveau à travers la barrière hémato-encéphalique, ont cessé en 1999 avec la découverte de l'enzyme sérine racémase dans le cerveau des rats par Herman Wolosker et al[85]. La sérine racémase catalyse la racémisation de la sérine. On ne connaissait précédemment les racémases d'acides aminés que chez les bactéries et quelques insectes. L'enzyme a été trouvée dans les cellules gliales, qui montrent comparativement de hautes concentrations en D-sérine. Avec la découverte de la sérine racémase, on a pu montrer que ce métabolisme archaïque des acides aminés D a été conservé au cours de l'évolution jusqu'aux mammifères[85], et qu'il y exerce encore une fonction importante dans la neurotransmission - comme cela devrait apparaître dans l'avenir[86]. Il faut abandonner le dogme que les acides aminés D n'ont pas de fonction particulière chez les eucaryotes. On sait aujourd'hui que la D-sérine joue un rôle important dans de nombreux processus du système nerveux central, comme l'apprentissage et la mémoire, mais aussi dans des troubles psychiques[87], des neuropathies et des maladies neurodégénératives[88],[61],[89].
Importance physiologique
Acides aminés D libres
Jusqu'à la fin des années 1990, on est parti de l'hypothèse que les acides aminés D n'avaient aucune fonction physiologique chez les vertébrés. Avec la découverte de relativement grandes quantités de D-sérine et de D-aspartate dans le cerveau des mammifères, l'étude de l'action physiologique de ces deux acides aminés inhabituels a commencé. Cette étude est une discipline relativement jeune, avec encore beaucoup de questions ouvertes.
La D-sérine
La D-sérine se trouve outre les cellules gliales également dans les neurones[91],[92]. Elle provient de la L-sérine sous l'action catalytique de l'enzyme sérine racémase (EC 5.1.1.18), qui est exprimée par ces cellules. La destruction est catalysée par l'oxydase des acides aminés D (EC 1.4.3.3). La concentration de D-sérine est déterminée par ces deux processus de formation et de destruction. La D-sérine est un co-agoniste du récepteur NMDA, dont le ligand naturel est la glycine[84]. Ce récepteur est de grande importance pour une série de processus physiologiques, et aussi pathologiques. La D-sérine renforce l'activité du récepteur NMDA. C'est pourquoi elle est aussi nommée neuromodulateur[93].
Une surexpression d'oxydase d'acides aminés D, conduisant à une augmentation de la dégradation de la D-sérine, réduit en conséquence l'activité des récepteurs NMDA. Cette activité diminuée est mise en relation avant tout avec la schizophrénie[94]. Déjà de faibles quantités d'antagonistes du récepteur peuvent déclencher chez des sujets sains des symptômes comme de faibles troubles cognitifs et physiologiques, qui correspondent à ceux d'une schizophrénie[95].
En 2002, un grand groupe de recherche international a établi que le gène nouvellement découvert G72 (gène DAOA, activateur de l'oxydase des acides aminés) est en lien étroit avec la schizophrénie. Le produit de G72 active l'oxydase des acides aminés D, ce qui fait diminuer la concentration de D-sérine dans le cerveau. Mais ils n'ont trouvé qu'une faible corrélation entre l'activité de l'oxydase des acides aminés D et la survenue de la schizophrénie. C'est pourquoi la combinaison de l'oxydase des acides aminés D et de l'activateur G72 les soutenait mutuellement (synergie)[96]. Les auteurs en ont conclu que finalement la concentration en D-sérine libre joue un rôle notable dans la schizophrénie. D'autres études ont montré également une relation génétique entre l'oxydase des acides aminés D et la schizophrénie[97],[98]. Ces découvertes s'accordent avec les résultats de groupes qui ont pu montrer que la concentration de D-sérine dans le sérum sanguin[99] et dans le liquide cérébrospinal[100],[101] de patients schizophréniques, par comparaison avec une cohorte de sujets sains, est significativement réduite. En outre, on a trouvé dans les cerveaux de patients schizophréniques décédés une expression plus élevée de l'oxydase des acides aminés D[102],[103],[104],[105]. L'administration en supplément de D-sérine pendant le traitement de patients schizophréniques a présenté dans des études cliniques de nombreux résultats encourageants[106],[107]. Dans une méta-analyse de 18 études cliniques, on a établi la réduction des symptômes de la schizophrénie. Cependant, l'amélioration n'était que modérée[108].
Les découvertes sur la fonction de la D-sérine et de l'oxydase des acides aminés D ont conduit à la mise au point de divers inhibiteurs de l'oxydase des acides aminés D, qui pourraient être des médicaments potentiels pour le traitemement de la schizophrénie[109],[110],[111]. Les inhibiteurs de l'oxydase des acides aminés D se trouvent encore dans une phase de mise au point très précoce[112],[113], si bien qu'en 2012, aucun médicament sur ce principe n'a encore reçu d'autorisation de mise sur le marché (AMM).
On étudie, dans l'autre sens, si une concentration trop élevée de ces acides aminés dans les cellules gliales, et l'excitotoxicité allant de pair, peut être une cause de la sclérose latérale amyotrophique, une maladie dégénérative du système nerveux[114],[115].
Le D-aspartate
C'est en 1986 que le groupe autour de l'Américain David S. Dunlop a trouvé des quantités substantielles de D-aspartate dans le cerveau de rongeurs et dans le sang humain. C'est dans le télencéphale de rats nouveau-nés qu'ils ont trouvé la plus haute concentration, avec 164 nmol/g de D-aspartate. Ceci correspondait à 8,4 % de l'aspartate total. Cette concentration dépasse celle de nombreux acides aminés L essentiels dans le cerveau[116]. En dehors du cerveau, ils ont pu aussi mettre en évidence des concentrations relativement élevées de D-aspartate dans la glande pinéale, l'hypophyse, les surrénales et les testicules[117],[118]. De manière analogue à la D-sérine, le D-aspartate est produit dans l'organisme par racémisation enzymatique de L-aspartate, et dans ce cas par la racémase du D-aspartate (EC 5.1.1.13), et la dégradation est produite par l'oxydase du D-aspartate (EC 1.4.3.1). Avec l'âge, la concentration de D-aspartate diminue drastiquement[119]. Les hautes activités en racémase du D-aspartate se trouvent dans les organes où on trouve également de hautes concentrations en D-aspartate. L'activité est maximale dans l'hypophyse. Une déactivation de la racémase du D-aspartate, par exemple par un rétrovirus, qui suscite une perte de fonction ciblée dans l'acide ribonucléique (ARN) complémentaire de la racémase du D-aspartate, conduit à une diminution substantielle de la concentration en D-aspartate. La conséquence en est que le développement dendritique est massivement perturbé, ce qui conduit alors à des dommages marqués pour la neurogénèse dans l'hippocampe[120]. Sur la base de ces résultats expérimentaux, on part de l'hypothèse que le D-aspartate est un régulateur important du développement neuronal[119]. L'action physiologique détaillée du D-aspartate est encore largement inconnue. Ce domaine de recherche est tout à fait nouveau. C'est ainsi que ce n'est qu'en 2010 que la racémase de l'aspartate a été clonée chez les mammifères[120].
Peptides contenant des acides aminés D
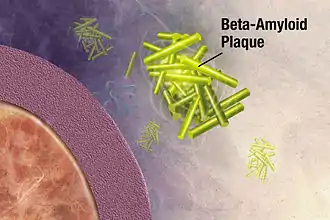
Au cours du vieillissement d'un organisme, la multiplication de la racémisation, en particulier de l'aspartate, amène une perte croissante d'homochiralité. Le stress oxydant et les rayons UV[121] peuvent accélérer cette perte. La racémisation de l'aspartate se déroule particulièrement facilement en raison de la formation d'un intermédiaire, un succinimide, qui n'a besoin que d'une très faible énergie d'activation[122]. Cette racémisation in vivo des protéines non enzymatique est un processus de vieillissement autonome, qui concerne avant tout les protéines à longue vie, comme le collagène de la dentine ou le cristallin[123]. Par exemple, 0,14 % de l'aspartate du cristallin est racémisé par an. Un homme de 30 ans a ainsi en moyenne 4,2 % de l'aspartate dans son cristallin qui est racémisé[124]. En outre d'autres protéines fonctionnelles, comme des enzymes ou des sémiochimiques, sont aussi touchées par la racémisation. Les peptides qui contiennent des acides aminés D sont notablement plus stables vis-à-vis de la dégradation enzymatique par des protéases que ceux dont les acides aminés ne sont qu'en conformation L. Dans bien des cas, la racémisation d'une protéine endogène conduit à des problèmes physiologiques. La racémisation conduit à une perte de fonction et à une accumulation de la protéine dans les tissus les plus divers, où l'organisme ne peut pas les dégrader. Dans certains tableaux cliniques, on observe un accroissement de la racémisation. Dans l'athérome, l'emphysème pulmonaire, la presbytie, la cataracte, les manifestations dégénératives des cartilages et du cerveau, on considère la racémisation de l'aspartate comme un facteur pathologique pertinent[125].
C'est en 1988 que l'on a trouvé pour la première fois dans les plaques séniles de bêta-amyloïde du cerveau de patients décédés avec la maladie d'Alzheimer un taux élevé de racémisation[126]. C'étaient avant tout du D-aspartate et de la D-sérine qui ont été mis en évidence[127]. Plus tard, on a reconnu qu'une racémisation de l'aspartate en position 23[128] conduisait à une accélération de l'agrégation des peptides[129] qui est considérée comme un élément substantiel de la pathogenèse de la maladie d'Alzheimer. Contrairement à la racémisation en position 23, la racémisation en position 7 conduit à une diminution de l'agrégation des peptides[129]. Dans la survenue de la maladie d'Alzheimer, on attribue un rôle important aux processus de racémisation de la bêta-amyloïde par veillissement de la protéine, qui se déroulent comme ceux de la dentine. La racémisation accélère l'agrégation des peptides et rend plus difficile leur désagrégation par des protéases[130],[131].
Propriétés
Propriétés chimiques et physiques
Dans un environnement achiral, les acides aminés L et D sont identiques dans leurs propriétés chimiques et physiques, à l'exception de leur action sur la direction de polarisation de la lumière. Par contre, dans un environnement chiral, des différences notables peuvent être établies. Ceci est vrai en particulier dans les processus biochimiques, qui sont naturellement chiraux. Un exemple pratique pour cela est la différence de goût entre des énantiomères d'acides aminés. Les chémorécepteurs gustatifs construits d'acides aminés L forment un environnement chiral, qui interagissent différemment vis-à-vis des énantiomères. C'est ainsi que le goût de la plupart des acides aminés L est décrit comme amer, tandis que celui des acides aminés D l'est comme doux[132],[133]. Un exemple extrême est le D-tryptophane, le plus sucré de tous les acides aminés D, plus de 37 fois plus que le saccharose. Par contre, le L-tryptophane, est, avec la L-tyrosine, le plus amer[134]. De la même manière, les interactions avec d'autres récepteurs ou enzymes peuvent se dérouler de façon différente dans les processus biochimiques. Ceci est en particulier valable pour les peptides et les protéines qui contiennent un ou plusieurs acides aminés D.
L'inclusion d'un acide aminé D, ou en particulier l'épimérisation d'un acide aminé L au sein d'une protéine produit du point de vue stéréochimique la formation d'un diastéréoisomère, qui donne à l'ensemble de la protéine des propriétés chimiques et physiques complètement nouvelles[31]. Cette intervention biochimique sur la structure primaire du peptide a des conséquences notables sur ses structures secondaire, tertiaire et quaternaire. Son effet biochimique est alors fortement modifié. Dans les cas extrêmes, elle peut soit perdre complètement sa fonction, soit prendre une fonction totalement nouvelle, éventuellement toxique. Les acides aminés D empêchent la constitution d'une hélice alpha au sein d'un peptide constitué d'acides aminés L. Ce sont des briseurs d'hélice. Seules des protéines constituées entièrement soit d'acides aminés D soit d'acides aminés L peuvent construire des hélices avec les acides aminés susceptibles de les construire (valine, glutamine, isoleucine, alanine, méthionine, leucine, glutamate ou tryptophane). Ces hélices tournent en sens inverse. Ceci n'est pas possible avec un mélange d'acides aminés de conformations différentes[14].
Toxicologie
Isomères D d'acides aminés protéinogènes

Dans des études où la prise orale d'acides aminés, par exemple sous la forme de compléments nutritifs, a été étudiée, il a été montré que tous les acides aminés sauf la sérine et l'aspartate ont des effets toxiques plus marqués en configuration L « naturelle » qu'en configuration D[135],[26]. Les acides aminés D forment une partie normale d'un nombre d'aliments. Ils ont pour origine avant tout des processus de racémisation d'acides aminés L. Dans les nourritures qui ont subi une fermentation, comme les produits lactés, on trouve des quantités élevées d'acides aminés D. Par exemple, l'emmental contient environ 0,7 g/kg d'acides aminés D[136]. Déjà dans le lait de vache frais, environ 1,5 % des acides aminés sont en configuration D[137].
On estime qu'environ un tiers des acides aminés D pris dans la nourriture sont d'origine microbienne[138]. Pour pouvoir utiliser dans l'organisme les acides aminés contenus dans la nourriture et insérés dans les protéines, les protéines doivent être décomposées dans leurs éléments, les acides aminés libres. S'il se trouve dans une protéine des acides aminés D, l'accès de la protéine aux enzymes protéolytiques peut être notablement limité. Les enzymes de l'appareil digestif humain ne peuvent pas briser les liaisons entre acides aminés D et L. La dégradation en acides aminés libres, ou en dipeptides ou tripeptides, nécessaire pour l'absorption dans la muqueuse intestinale en est rendue plus difficile[139]. Les parties plus importantes de peptides ne peuvent pas être assimilées et sont excrétées avec les matières fécales. La disponibilité biologique, et également la valeur nutritive est alors fortement diminuée[140]. Les dipeptides ou tripeptides contenant des acides aminés D, et les acides aminés D libres peuvent être résorbés par des transporteurs de peptides. Une grande partie des acides aminés D ainsi absorbés sont éliminés par les reins. Selon l'offre de nourriture et le type d'acide aminé, une partie des acides aminés D peut être transformée par épimérisation en acides aminés L, et donc être mise à la disposition de la biosynthèse des protéines[141].
L'inclusion d'acides aminés D dans la paroi cellulaire des bactéries a pour effet leur résistance aux protéases. Cette résistance est très importante pour l'homme, puisqu'il y a dans l'intestin d'un adulte plusieurs centaines de grammes de bactéries intestinales, qui sont nécessaires à la digestion, avec une multiplicité de protéases.
La plus grande partie des acides aminés D dans la nourriture trouve son origine par sa préparation. Les hautes températures ou des conditions fortement acides ou basiques conduisent à une racémisation partielle. Par exemple, dans les chips, environ 14 % de l'aspartate est sous forme D ; dans l'ersatz de lait à mettre dans le café, ce sont 17 %, et dans une tranche de bacon pour le petit-déjeuner, 13 %. Les acides aminés L libres se racémisent environ dix fois plus lentement que s'ils sont liés dans une protéine. Le taux de racémisation en outre dépend fortement de l'acide aminé. Par exemple, la sérine, en raison de son groupe hydroxyle se racémise particulièrement facilement[140],[142],[143]. Les conditions drastiques nécessitées par la production de gélatine - fusion acide ou basique à haute température - conduisent à une forte racémisation, en particulier de l'aspartate, dans le collagène de la gélatine. La fraction de D-aspartate sur aspartate total peut facilement dépasser 30 % dans des gélatines disponibles dans le commerce[144].

Les acides aminés D pris par les mammifères ne sont pas incorporés dans des protéines ou des peptides, ou d'autres (macro-)molécules du métabolisme. On ne constate pas d'enrichissement des tissus corporels. Les acides aminés D sont éliminés en partie dans l'urine, et en partie par désamination par l'enzyme oxydase des acides aminés D présente dans le foie et les reins, et oxydés en produits normaux du métabolisme, les cétoacides. En ce qui concerne la toxicité des infusions d'acides aminés D, on possède de longues années d'expérience plus ou moins volontaire, qui amènent à conclure qu'ils ne sont pas nuisibles à la santé[145]. La base pour cette affirmation est la bonne tolérance de l'alimentation par voie parentérale qui a consisté pendant de nombreuses années de racémates d'acides aminés à haute dose. Ces solutions d'infusion étaient obtenues par hydrolyse acide de protéines, ce qui conduit forcément à une forte racémisation[146]. La méthionine racémique, DL-méthionine, constitue une partie de beaucoup d'aliments de l'élevage bovin. Chez les vaches laitières, on a pu montrer que plus de 75 % de la D-méthionine était transformée en L-méthionine, et ainsi rendue biodisponible[147].
Indépendamment de ces valeurs tirées de l'usage, il faut considérer les résultats expérimentaux sur le modèle animal du rat. De hautes doses (dans la gamme des 0,8 g/kg) de D-sérine conduisent dans ces organismes modèles à une nécrose tubulaire aiguë[148],[149],[150],[151], réversible après la suppression de l'administration de D-sérine[152]. Au bout d'environ six jours, la régénération complète de la fonction du rein est acquise[153]. Les modifications pathologiques sont largement similaires à une lésion des reins due à de la lysinoalanine (combinaison de lysine et de déshydroalanine, un acide aminé non protéinogène). On n'a pas encore d'explication sûre de la raison pour laquelle la D-sérine à ces hautes concentrations est toxique pour le rein. Il est possible que la D-sérine réduise la concentration en glutathion rénal, dont la fonction est de protéger les cellules du tubule distal des influences néfastes dus aux dérivés réactifs de l'oxygène (ROS). Dans la dégradation enzymatique de la D-sérine par l'oxydase des acides aminés D, il se forme comme sous-produit du peroxyde d'hydrogène[154], qui abaisse notablement la réserve de glutathion au sein de la cellule[140],[155].

Une communication publiée en décembre 1989 dans le périodique spécialisé de renom The Lancet a suscité une grande attention. Elle était signée de trois médecins viennois, qui avaient chauffé du lait dans un four à micro-ondes, et trouvé de grandes quantités de D-proline, apparemment due à la racémisation de la L-proline du lait. À la suite, ils ont attribué à cette D-proline des propriétés toxiques pour les nerfs, les reins et le foie[156]. Cette publication était une lettre aux rédacteurs, et non une publication soumise à critique par les pairs, ni même à une étude contrôlée[157]. Les auteurs n'ont pas précisé les conditions de l'expérience qui avait conduit à ce point de racémisation. Indépendamment de ces restrictions, cette annonce a été publiée dans la presse quotidienne et hebdomadaire avec des formules dramatiques et des avertissements sur l'usage des fours à micro-ondes. En , le bureau fédéral de la santé a publié une mise au point, qui n'a pratiquement eu aucun impact dans le public. D'autres scientifiques ont souligné que la D-proline fait normalement partie de la nourriture quotidienne, et qu'elle est vite dégradée et éliminée après son absorption orale[158]. Cependant, en , un journal est paru avec la manchette « Les micro-ondes empoisonnent les nerfs, le foie et les reins[146]. » Des affirmations semblables se trouvent toujours aujourd'hui sur des sites web correspondants[159],[160],[161].
Les tentatives d'autres groupes pour retrouver les résultats des médecins viennois ont commencé par échouer. Ainsi, après 30 minutes de cuisson du lait sur une cuisinière, aucune augmentation de la D-proline n'a pu être mesurée[162],[163]. Deux ans plus tard, les conditions de l'expérience ont été publiées. Les auteurs de la lettre au Lancet avaient chauffé le lait dans un récipient à pression fermé pendant 10 minutes à 174−176 °C, un domaine de température que les récipients domestiques pour le chauffage du lait ne peuvent pas atteindre[164],[165]. En ce qui concerne leur affirmation sur la neurotoxicité de la D-proline, les auteurs de la lettre au Lancet se fondaient sur des expériences de 1978, où l'on avait injecté la substance directement dans les ventricules cérébraux[166]. Des expériences ultérieures sur la toxicité de la D-proline chez les rats ont montré que ce composé est sans danger, même à haute concentration[167],[146].
Le vrai danger du chauffage du lait dans les fours à micro-ondes provient - spécialement pour les enfants en bas âge - du chauffage inégal du contenu du biberon, ce qui conduit couramment à des brûlures de gravité clinique[168].
Isomères D d'acides aminés non protéinogènes
On ne peut pas donner d'indication générale sur la toxicité des isomères D des acides aminés non-protéinogènes. Elle dépend très individuellement du type d'acide aminé concerné. De façon intéressante, certaines combinaisons contenant des acides aminés D sont notablement moins toxiques que leurs isomères L : par exemple, la cyclosérine et la pénicillamine. C'est ainsi que par exemple, la dose létale médiane pour l'absorption orale du racémique de pénicillamine D et L chez le rat est de 365 mg/kg. Pour la D-pénicillamine, on ne note aucun signe de toxicité même pour une dose de 1 200 mg/kg[169].
D-Peptides
On ne peut pas faire d'assertion générale sur les propriétés toxicologiques des D-peptides. Leur sensibilité vis-à-vis des protéases est notablement plus faible et leur potentiel immunogène significativement plus petit que celui des L-peptides correspondants[170],[171].
Analyse
Procédés classiques

Avec un polarimètre, on peut déterminer l'angle de rotation de la polarisation d'une solution d'acides aminés, d'où l'on peut calculer le contenu en énantiomères D et L. Pour cela, il faut que des conditions standardisées soient réalisées (avant tout, concentration, température et solvant). En outre, ce procédé n'est applicable qu'à des acides aminés seuls, et non à des mélanges d'acides aminés différents. Dans les années 1960 à 1980, la chromatographie à échange d'ions a été utilisée pour la séparation d'acides aminés dérivés. Pour cela, les acides aminés à analyser sont transformés avant la séparation en dipeptides diastéréoisomères avec des acides aminés L[172]. Également des procédés enzymatiques, qui reposent sur des transformations avec des enzymes spécifiques comme les oxydases des acides aminés D ou L[173] font partie des procédés classiques de détermination des énantiomères des acides aminés[26].
Procédés chromatographiques
Des analyses quantitatives, même de mélanges complexes d'acides aminés, peuvent être accomplies au moyen de procédés chromatographiques. Ceci suppose de séparer les composants individuels du mélange sur la phase stationnaire du chromatographe, puis de les mesurer avec un détecteur. Ces derniers sont surtout des spectroscopes ou des spectomètres de masse, ou encore en chromatographie en phase gazeuse par détection d'ionisation en flamme (en). Pour la séparation du mélange sur la phase stationnaire, on utilise deux stratégies. Dans le cas le plus simple, cette séparation des deux énantiomères a lieu sur une phase stationnaire chirale, qui interagit différemment avec les deux isomères, et donc est éluée à une vitesse différente. La séparation à une phase stationnaire achirale n'est possible que quand les énantiomères sont remplacés par des diastéréoisomères. Les procédés d'analyse qui se sont imposés sont la chromatographie en phase gazeuse (GC) et la chromatographie en phase liquide à haute performance (HPLC). Comme procédé non-chromatographique, on utilise notamment l'électrophorèse capillaire pour l'analyse des acides aminés D[26]. Ce n'est que la mise au point de méthodes chromatographiques spéciales qui a permis de trouver et de quantifier les acides aminés D dans les organes des organismes supérieurs[174],[175].
Chromatographie en phase gazeuse

Les acides aminés ne peuvent pas être vaporisés sans se décomposer. Pour leur séparation et leur analyse en chromatographie en phase gazeuse, ils doivent être transformés en composés vaporisables sans décomposition. À cette fin, ils sont en général soumis à une réaction chimique en deux étapes. Par exemple, dans une première étape, on peut transformer le groupe carboxyle en ester avec de l'éthanol, puis dans une deuxième étape, transformer le groupe amino en trifluoracétyle sous l'action de l'anhydride trifluoroacétique. Le dérivé N-TFA/O-éthyl- de l'acide aminé peut alors être vaporisé dans le chromatographe sans se décomposer, et être séparé sur une phase stationnaire chirale[176]. La réaction séparée sur chacun des ligands du carbone α protège du danger d'une racémisation, et garantit que la réaction aura la même cinétique pour les deux énantiomères. L'une ou l'autre de ces possibilités pourrait altérer le résultat de la mesure[26].
Chromatographie en phase liquide à haute performance

En HPLC, contrairement à la chromatographie en phase gazeuse, il est usuel de combiner le produit à analyser avec des réactifs chiraux, et d'utiliser une phase non-chirale. Comme réactif chiral, on peut utiliser la L-N-acétylcystéine avec du benzol-1,2-dicarbaldéhyde[177]. La paire de diastéréomères (D - L et L - L) ont des propriétés physiques et chimiques différentes, si bien qu'ils peuvent être séparés et détectés sur une colonne conventionnelle (achirale).
Synthèse

La plupart des acides aminés L protéinogènes sont obtenus par fermentation. Ce procédé microbiologique n'est pas approprié pour la production d'acides aminés D[178]. Pour couvrir les besoins croissants en acides aminés D, divers processus de production ont été mis au point.
Les synthèses chimiques classiques, comme la synthèse de Strecker conduisent toujours à des racémiques d'acides aminés. On peut séparer les énantiomères de ce mélange, ce qui est coûteux, ou bien on y transforme enzymatiquement les acides aminés L en cétoacides au moyen de désaminases d'acides aminés L, et on peut les séparer alors facilement[179],[180].
Une méthode plus élégante est la synthèse d'acides aminés D par la substitution d'hydantoïnes. Les hydantoïnes se produisent par des techniques de grands volumes selon la réaction de Bucherer-Bergs à partir d'aldéhydes, de cyanure de potassium et de carbonate d'ammonium. Selon le choix de l'aldéhyde utilisé, on obtiendra l'acide aminé souhaité. L'hydantoïne ainsi préparée peut ensuite être transformée par le procédé à l'hydantoïnase en acide aminé D. Ce procédé à plusieurs enzymes a été mis au point par Degussa (maintenant Evonik), et consiste en trois étapes. Tout d'abord, le dérivé d'hydantoïne racémique est hydrolysé sous l'influence catalytique d'une D-hydantoïnase en N-carbamoyl-D-acide aminé. Dans une deuxième étape, le N-carbamoyl-D-acide aminé est hydrolysé au moyen d'une D-carbamoylase en un acide aminé énantiomère pur. Dans la troisième étape, l'énantiomère non transformé du composé de l'hydantoïne est racémisé chimiquement ou enzymatiquement. La racémisation chimique a lieu pour un pH > 8, et peut être substantiellement accélérée par l'addition d'une racémase. En comparaison avec les autres processus, le processus à l'hydantoïne produit à partir d'un racémique des acides aminés énantiomères purs avec un rendement théorique de 100 %[181].
Utilisation
La demande mondiale en acides aminés D a continuellement augmenté au cours des dernières années. Pour l'année 2017, on pronostique un marché d'environ 3,7 milliards de US $[182].
Les acides aminés D sont utilisés comme constituants importants par exemple dans des édulcorants, des insecticides, des cosmétiques et avant tout dans une multiplicité de médicaments, qui représentent un facteur de croissance important pour le développement du marché[183].
C'est ainsi que chaque année, on a besoin de plusieurs milliers de tonnes de D-4-hydroxyphénylglycine et de D-phénylglycine pour la synthèse des pénicillines (par exemple l'amoxicilline) et des céphalosporines (par exemple le Céfaclor).
Non seulement les acides aminés D élèvent la stabilité des parois cellulaires des bactéries vis-à-vis de la dégradation protéolytique, mais leur inclusion bien dirigée dans les médicaments améliore leur stabilité, en particulier face à l'administration orale. En outre, le changement de l'ordonnancement des groupes fonctionnels dans leur conformation offre pour la construction de nouvelles molécules un nouveau degré de liberté qui peut conduire à de meilleures propriétés[184]. Le cetrorelix (en), utilisé en médecine de l'appareil génital, pour inhiber l'hormone de libération des gonadotrophines hypophysaires, dont il est un analogue, consiste par exemple en dix acides aminés, dont cinq sont en configuration D[185],[186]. Le cetrorelix est entièrement fabriqué synthétiquement à partir des acides aminés individuels. D'autres médicaments similaires, comme leuproreline, buserelin, degarelix, histrelin, nafarelin ou abarelix contiennent au moins un acide aminé D.
Le tadalafil, utilisé dans le traitement de la dysfonction érectile, mieux connu sous sa marque commerciale Cialis, contient dans sa structure un D-tryptophane[187]. L'antidiabétique natéglinide, du groupe des glinides est fait de D-phénylalanine et d'acide cis-4-isopropyl-cyclohexane-carbonique. La phénylalanine est utilisée depuis les années 1970 comme antidépresseur[188]. Pour l'utilisation comme médicament, on utilise le racémique, bien moins onéreux. Une fraction substantielle de l'action antidépressive et analgésique est due à la D-phénylalanine, qui n'est pas métabolisée, comme la L-phénylalanine en L-tyrosine, L-DOPA ou noradrénaline qui améliorent l'humeur, mais elle bloque de façon primaire l'enzyme enképhalinase[189]. Ce blocage conduit à augmenter la concentration d'enképhalines dans le sang, ce qui suscite l'effet analgésique. Ultérieurement, la D-phénylalanine est métabolisée principalement en phényléthylamine[190],[191]
L'insecticide fluvalinate utilisé pout la lutte contre le varroa[192] du groupe des pyréthrinoïdes contient dans sa composition de la D-valine.
La D-alanine est un composant de l'édulcorant alitame.
Pour aller plus loin
- (de) Hans-Dieter Belitz, Werner Grosch et Peter Schieberle, Lehrbuch der Lebensmittelchemie, Springer Verlag, , 5e éd. (ISBN 3-540-41096-1, lire en ligne)
- (en) Ryuichi Konno, Hans Brückner, Antimo D'Aniello, George Fisher, Noriko Fujii et Hiroshi Homma, D-amino acids: a new frontier in amino acids and protein research – practical methods and protocols., Nova Science Publishers, , 629 p. (ISBN 1-600-21075-9)
- (en) Loredano Pollegioni et Stefano Servi (dir.), Unnatural Amino Acids, Humana Press, , 409 p. (ISBN 1-617-79330-2)
- (en) Gyula Pályi, Luciano Caglioti et Claudia Zucchi (dir.), Advances in BioChirality, Elsevier, (ISBN 0-080-43404-5, Modèle:Google Buch)
Liens web
- (de) « Die D- und die L-Form der Aminosäuren » (consulté le )
- (de) Hanka Symmank, « Funktionelle und strukturelle Charakterisierung bakterieller Peptidsynthetasen. », sur Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, (consulté le )
Notes et références
- Belitz2001, p. 15.
- (en) Uwe Meierhenrich, Amino Acids and the Asymmetry of Life: Caught in the Act of Formation, Springer, (ISBN 3-540-76885-8, lire en ligne), p. 47, 53–54.
- (en) V. S. Lamzin, Z. Dauter et K. S. Wilson, « How nature deals with stereoisomers », Current opinion in structural biology, vol. 5, no 6, , p. 830–836 (ISSN 0959-440X, PMID 8749373).
- (en) S. A. Fuchs, R. Berger et al., « D-amino acids in the central nervous system in health and disease », Molecular genetics and metabolism, vol. 85, no 3, , p. 168–180 (ISSN 1096-7192, PMID 15979028, DOI 10.1016/j.ymgme.2005.03.003) (Review)
- (en) G. F. Joyce, G. M. Visser et al., « Chiral selection in poly(C)-directed synthesis of oligo(G) », Nature, vol. 310, no 5978, , p. 602–604 (ISSN 0028-0836, PMID 6462250)
- (en) V. V. Avetisov et V. I. Goldanskii, « Homochirality and stereospecific activity: evolutionary aspects », Bio Systems, vol. 25, no 3, , p. 141–149 (ISSN 0303-2647, PMID 1912384).
- (en) W. A. Bonner, « Experimental evidence for beta-decay as a source of chirality by enantiomer analysis », Origins of life, vol. 14, nos 1–4, , p. 383–390 (ISSN 0302-1688, PMID 11536584) (Review)
- (en) W. A. Bonner, « Parity violation and the evolution of biomolecular homochirality », Chirality, vol. 12, no 3, , p. 114–126 (ISSN 0899-0042, PMID 10689289, DOI 10.1002/(SICI)1520-636X(2000)12:3<114::AID-CHIR3>3.0.CO;2-N) (Review)
- (en) J. R. Cronin et S. Pizzarello, « Enantiomeric excesses in meteoritic amino acids », Science, vol. 275, no 5302, , p. 951–955 (ISSN 0036-8075, PMID 9020072).
- (en) S. Pizzarello, M. Zolensky et K. A. Turk, « Nonracemic isovaline in the Murchison meteorite: chiral distribution and mineral association », Geochimica et Cosmochimica Acta, vol. 67, no 8, , p. 1589–1595 (DOI 10.1016/S0016-7037(02)01283-8)
- (en) P. Schmitt-Kopplin, Z. Gabelica et al., « High molecular diversity of extraterrestrial organic matter in Murchison meteorite revealed 40 years after its fall », PNAS, vol. 107, no 7, , p. 2763–2768 (ISSN 1091-6490, PMID 20160129, PMCID 2840304, DOI 10.1073/pnas.0912157107).
- (en) D. P. Glavin et J. P. Dworkin, « Enrichment of the amino acid L-isovaline by aqueous alteration on CI and CM meteorite parent bodies », PNAS, vol. 106, no 14, , p. 5487–5492 (ISSN 1091-6490, PMID 19289826, PMCID 2667035, DOI 10.1073/pnas.0811618106).
- (en) P. W. Lucas, J. H. Hough et al., « UV circular polarisation in star formation regions: the origin of homochirality? », Origins of life and evolution of the biosphere, vol. 35, no 1, , p. 29–60 (ISSN 0169-6149, PMID 15889649).
- (de) Thomas Carell, « Vorlesung Stereochemie : chap. 9 : Racemisierungen », sur LMU München (consulté le ), p. 150.
- (de) Elizabeth R. Neswald, Thermodynamik als kultureller Kampfplatz: zur Faszinationsgeschichte der Entropie, 1850–1915, Rombach, (ISBN 3-793-09448-0), p. 335.
- (de) A. S. Kekulé, « Ach wie gut, dass niemand weiß … », Tagesspiegel, (lire en ligne).
- (en) T. Ogino et H. Ogino, « Application to forensic odontology of aspartic acid racemization in unerupted and supernumerary teeth », Journal of dental research, vol. 67, no 10, , p. 1319–1322 (ISSN 0022-0345, PMID 3170888).
- (en) T. Ogino, H. Ogino et B. Nagy, « Application of aspartic acid racemization to forensic odontology: post mortem designation of age at death », Forensic science international, vol. 29, nos 3–4, , p. 259–267 (ISSN 0379-0738, PMID 4076954)
- (en) S. Ohtani et T. Yamamoto, « Strategy for the estimation of chronological age using the aspartic acid racemization method with special reference to coefficient of correlation between D/L ratios and ages », Journal of forensic sciences, vol. 50, no 5, , p. 1020–1027 (ISSN 0022-1198, PMID 16225206) (Review)
- (en) J. L. Bada, B. Herrmann et al., « Amino acid racemization in bone and the boiling of the German Emperor Lothar I », Applied Geochemistry, vol. 4, no 3, , p. 325–327 (DOI 10.1016/0883-2927(89)90036-X).
- (en) Chris McManus, Right Hand, Left Hand – The Origins of Asymmetry in Brains, Bodies, Atoms and Cultures, Harvard University Press, (ISBN 0-674-01613-0, lire en ligne), p. 130-132.
- (en) P. M. Masters et M. Friedman, « Racemization of amino acids in alkali-treated food proteins », Journal of agricultural and food chemistry, vol. 27, no 3, , p. 507–511 (ISSN 0021-8561, PMID 447924).
- (en) J. L. Bada, « Kinetics of racemization of amino acids as a function of pH », Journal of the American Chemical Society, vol. 94, no 4, , p. 1371–1373 (ISSN 0002-7863, PMID 5060280).
- (en) H. Frank, W. Woiwode et al., « Determination of the rate of acidic catalyzed racemization of protein amino acids », Liebigs Ann Chem., no 3, , p. 354–365 (DOI 10.1002/jlac.198119810303).
- (en) T. Geiger et S. Clarke, « Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation », The Journal of biological chemistry, vol. 262, no 2, , p. 785–794 (ISSN 0021-9258, PMID 3805008)
- (de) Thorsten Erbe (Dissertation), « Die Quantifizierung von Aminosäurenisomeren in Lebensmitteln mittels chiraler Gaschromatographie-Massenspektrometrie im Hinblick auf die Relevanz und die Entstehungsmechanismen von D-Aminosäuren », sur Justus-Liebig-Universität Gießen, (consulté le ).
- (en) A. Paquet et M. Ching-Yung, « Racemization assessment in alkali treated dietary proteins using high-performance liquid chromatography », Nutrition Research, vol. 9, no 9, , p. 1053–1065 (DOI 10.1016/S0271-5317(89)80066-1).
- (en) M. Friedman, « Chemistry, nutrition, and microbiology of D-amino acids », Journal of agricultural and food chemistry, vol. 47, no 9, , p. 3457–3479 (ISSN 0021-8561, PMID 10552672) (Review)
- (en) J. P. Richard et T. L. Amyes, « Proton transfer at carbon », Current opinion in chemical biology, vol. 5, no 6, , p. 626–633 (ISSN 1367-5931, PMID 11738171) (Review)
- (en) J. P. Richard, et T. L. Amyes, « On the importance of being zwitterionic: enzymatic catalysis of decarboxylation and deprotonation of cationic carbon », Bioorganic chemistry, vol. 32, no 5, , p. 354–366 (ISSN 0045-2068, PMID 15381401, DOI 10.1016/j.bioorg.2004.05.002) (Review)
- (de) Daniel Björn Stein, « Substratspezifität und Funktionalität von Epimerisierungsdomänen in der nichtribosomalen Peptidsynthese », Dissertation, sur Philipps-Universität Marburg, (consulté le ), p. 29.
- (en) S. Glavas et M. E. Tanner, « Active site residues of glutamate racemase », Biochemistry, vol. 40, no 21, , p. 6199–6204 (ISSN 0006-2960, PMID 11371180).
- (en) L. M. Fisher, J. G. Belasco et al., « Energetics of proline racemase: transition-state fractionation factors for the two protons involved in the catalytic steps », Biochemistry, vol. 25, no 9, , p. 2543–2551 (ISSN 0006-2960, PMID 3521738).
- (en) Geoffrey Zubay, Origins of Life: On Earth and in the Cosmos, Academic Press, (ISBN 0-127-81910-X, lire en ligne), p. 296
- (de) Emil Abderhalden, Fermentforschung, t. 16–17, S. Hirzel, , p. 301
- (de) H. A. Krebs, « Untersuchungen über den Stoffwechsel der Aminosäuren im Tierkörper », Hoppe-Seyler's Zeitschrift für physiologische Chemie, vol. 217, , p. 191
- (en) H. A. Krebs, « Metabolism of amino-acids: Deamination of amino-acids », The Biochemical journal, vol. 29, no 7, , p. 1620–1644 (ISSN 0264-6021, PMID 16745832, PMCID 1266672)
- (en) H. Blaschko et J. Hawkins, « D-Amino acid oxidase in the molluscan liver », The Biochemical journal, vol. 52, no 2, , p. 306–310 (ISSN 0264-6021, PMID 13018226, PMCID 1197987)
- (de) Felix Ehrlich, « Über asymmetrische und symmetrische Einwirkung von Hefe auf Racemverbindungen natürlich vorkommender Aminosäuren », Biochem Z., vol. 63, , p. 379–401
- (de) Edmund Oskar von Lippmann, « Ueber das Vorkommen von Leucin und Tyrosin in der Rübenmelasse », Ber Dtsch Chem Ges., vol. 17, , p. 2835–2840 (DOI 10.1002/cber.188401702243)
- (de) S. Fränkel, H. Gallia, A. Liebster et S. Rosen, « Über die Produkte prolongierter tryptischer Verdauung des Caseins », Biochem Z., vol. 145, , p. 225–241
- (de) E. Winterstein, C. Reuter et R. Korolew, « Ueber die chemische Zusammensetzung einiger Pilze und über die bei der Autolyse derselben auftretenden Produkte », Landw Versuchsstat., vol. LXXIX - LXXX, , p. 541–562
- (en) J. H. Birkinshaw, H. Raistrick et G. Smith, « Studies in the biochemistry of micro-organisms: Fumaryl-dl-alanine (fumaromono-dl-alanide), a metabolic product of Penicillium resticulosum sp.nov. », The Biochemical journal, vol. 36, nos 10–12, , p. 829–835 (ISSN 0264-6021, PMID 16747516, PMCID 1266878)
- (en) T. Robinson, « D-amino acids in higher plants », Life sciences, vol. 19, no 8, , p. 1097–1102 (ISSN 0024-3205, PMID 792607) (Review)
- (en) J. L. Frahn et R. J. Illman, « The occurrence of D-alanine and D-alanyl-D-alanine in Phalaris tuberosa », Phytochem, vol. 14, , p. 1464–1465 (DOI 10.1016/S0031-9422(00)98674-6)
- (en) Y. Gogami, K. Ito et al., « Occurrence of D-serine in rice and characterization of rice serine racemase », Phytochemistry, vol. 70, no 3, , p. 380–387 (ISSN 0031-9422, PMID 19249065, DOI 10.1016/j.phytochem.2009.01.003)
- (en) T. Ogawa, M. Fukuda et K. Sasaoka, « Occurrence of N-malonyl-D-alanine in pea seedlings », Biochimica et biophysica acta, vol. 297, no 1, , p. 60–69 (ISSN 0006-3002, PMID 4144329)
- (en) H. Brückner, S. Haasmann et A. Friedrich, « Quantification of D-amino acids in human urine using GC-MS and HPLC », Amino Acids, vol. 6, , p. 205–211 (DOI 10.1007/BF00805848)
- (en) R. H. Buck et K. Krummen, « High-performance liquid chromatographic determination of enantiomeric amino acids and amino alcohols after derivatization with o-phthaldialdehyde and various chiral mercaptans. Application to peptide hydrolysates », Journal of chromatography, vol. 387, , p. 255–265 (ISSN 0021-9673, PMID 3558624).
- (en) E. E. Snell et B. M. Guirard, « Some Interrelationships of Pyridoxine, Alanine and Glycine in Their Effect on Certain Lactic Acid Bacteria », PNAS, vol. 29, no 2, , p. 66–73 (ISSN 0027-8424, PMID 16588604, PMCID 1078561)
- (en) J. Olivard et E. E. Snell, « Growth and enzymatic activities of vitamin B6 analogues. I. D-Alanine synthesis », The Journal of biological chemistry, vol. 213, no 1, , p. 203–214 (ISSN 0021-9258, PMID 14353919, PMCID 1078561, lire en ligne)
- (en) « D-amino acids in animals », Science, vol. 164, no 3876, , p. 142–149 (ISSN 0036-8075, PMID 5774186).
- (en) E. E. Snell, « The vitamin B6 group: VII. Replacement of vitamin B6 for some microorganisms by d(−)-alanine and an unidentified factor from casein », J Biol Chem., vol. 158, , p. 497–503 (lire en ligne)
- (de) Albert Gossauer, Struktur und Reaktivität der Biomoleküle, John Wiley & Sons, (ISBN 3-906-39029-2, lire en ligne), p. 347
- (en) W. A. Wood et I. C. Gunsalus, « D-Alanine formation; a racemase in Streptococcus faecalis », The Journal of biological chemistry, vol. 190, no 1, , p. 403–416 (ISSN 0021-9258, PMID 14841188)
- (en) J. Ju, H. Misono et K. Ohnishi, « Directed evolution of bacterial alanine racemases with higher expression level », Journal of bioscience and bioengineering, vol. 100, no 3, , p. 246–254 (ISSN 1389-1723, PMID 16243272, DOI 10.1263/jbb.100.246, lire en ligne)
- (en) R. J. Thompson, H. G. Bouwer et al., « Pathogenicity and immunogenicity of a Listeria monocytogenes strain that requires D-alanine for growth », Infection and immunity, vol. 66, no 8, , p. 3552–3561 (ISSN 0019-9567, PMID 9673233, PMCID 108386)
- (en) D. Billot-Klein, L. Gutmann et al., « Modification of peptidoglycan precursors is a common feature of the low-level vancomycin-resistant VANB-type Enterococcus D366 and of the naturally glycopeptide-resistant species Lactobacillus casei, Pediococcus pentosaceus, Leuconostoc mesenteroides, and Enterococcus gallinarum », Journal of bacteriology, vol. 176, no 8, , p. 2398–2405 (ISSN 0021-9193, PMID 8157610, PMCID 205365)
- (en) P. E. Reynolds, H. A. Snaith et al., « Analysis of peptidoglycan precursors in vancomycin-resistant Enterococcus gallinarum BM4174 », The Biochemical journal, vol. 301, , p. 5–8 (ISSN 0264-6021, PMID 8037690, PMCID 1137133)
- (en) C. A. Arias, M. Martín-Martinez et al., « Characterization and modelling of VanT: a novel, membrane-bound, serine racemase from vancomycin-resistant Enterococcus gallinarum BM4174 », Molecular microbiology, vol. 31, no 6, , p. 1653–1664 (ISSN 0950-382X, PMID 10209740)
- (de) Norma Christine Stäbler, « Untersuchungen zur Bildung von D-Aminosäuren mit Corynebacterium glutamicum », sur Dissertation, Heinrich-Heine-Universität Düsseldorf, (consulté le ), p. 7
- (en) M. F. Freeman, C. Gurgui et al. (Publication électronique avant impression), « Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides », Science (New York, N.Y.), (ISSN 1095-9203, PMID 22983711, DOI 10.1126/science.1226121)
- (en) T. Hamada, S. Matsunaga et al., « Solution structure of polytheonamide B, a highly cytotoxic nonribosomal polypeptide from marine sponge », Journal of the American Chemical Society, vol. 132, no 37, , p. 12941–12945 (ISSN 1520-5126, PMID 20795624, DOI 10.1021/ja104616z)
- (de) Dankwart Ackermann et M. Mohr, « Über stickstoffhaltige Bestandteile der Leber des Haifisches (Acanthias vulgaris) », Z Biol., vol. 98, no 37, , p. 26
- (en) L. R. Lyle et J. W. Jutila, « D-amino acid oxidase induction in the kidneys of germ-free mice », Journal of bacteriology, vol. 96, no 3, , p. 606–608 (ISSN 0021-9193, PMID 4389707, PMCID 252348)
- (en) J. L. Auclair et R. L. Patton, « On the occurrence of D-Alanine in the haemolymph of the milkweed bug, Oncopeltus fasciatus », Revue canadienne de biologie, vol. 9, no 1, , p. 3–8 (ISSN 0035-0915, PMID 15417891)
- (en) Gianluca Molla, Luciano Piubelli et al., « Enzymatic Detection of D-Amino Acids », dans Loredano Pollegioni, Stefano Servi, Unnatural Amino Acids, vol. 794, (ISBN 978-1-61779-330-1, DOI 10.1007/978-1-61779-331-8_18), p. 273–289
- (en) N. G. Srinivasan, J. J. Corrigan et A. Meister, « Biosynthesis of D-Serine in the silkworm, Bombyx mori », The Journal of biological chemistry, vol. 240, , p. 796–800 (ISSN 0021-9258, PMID 14275137, lire en ligne [PDF])
- (en) J. J. Corrigan et N. G. Srinivasan, « The occurrence of certain D-amino acids in insects », Biochemistry, vol. 5, no 4, , p. 1185–1190 (ISSN 0006-2960, PMID 5958195)
- (en) Y. Nagata, K. Yamamoto et al., « The presence of free D-alanine, D-proline and D-serine in mice », Biochimica et biophysica acta, vol. 1115, no 3, , p. 208–211 (ISSN 0006-3002, PMID 1346751)
- (en) P. Melchiorri et L. Negri, « The dermorphin peptide family », General pharmacology, vol. 27, no 7, , p. 1099–1107 (ISSN 0306-3623, PMID 8981054) (Review)
- (en) A. Anastasi, V. Erspamer et J. M. Cei, « Isolatioan and amino acid sequence of physalaemin, the main active polypeptide of the skin of Physalaemus fuscumaculatus », Archives of biochemistry and biophysics, vol. 108, , p. 341–348 (ISSN 0003-9861, PMID 14240587)
- (en) Rebecca Jo Jackway, « Biologically Active Peptides from Australian Amphibians », sur PhD Thesis, University of Adelaide, (consulté le ), p. 165
- (en) Alton Meister, Biochemistry of the amino acids, Academic Press, « At this time there is no conclusive evidence for the occurrence of D-amino acids in the proteins of plants and animals. (sic) »
- (en) M. Broccardo, V. Erspamer et al., « Pharmacological data on dermorphins, a new class of potent opioid peptides from amphibian skin », British journal of pharmacology, vol. 73, no 3, , p. 625–631 (ISSN 0007-1188, PMID 7195758, PMCID 2071698)
- (en) V. Erspamer, P. Melchiorri et al., « Deltorphins: a family of naturally occurring peptides with high affinity and selectivity for delta opioid binding sites », PNAS, vol. 86, no 13, , p. 5188–5192 (ISSN 0027-8424, PMID 2544892, PMCID 297583)
- (en) M. Amiche, A. Delfour et P. Nicolas, « Opioid peptides from frog skin. », dans Pierre Jollès, D-Amino Acids in Sequences of Secreted Peptides of Multicellular Organisms., Springer, (ISBN 3-764-35814-9, lire en ligne), p. 57–72
- (en) L. H. Lazarus et M. Attila, « The toad, ugly and venomous, wears yet a precious jewel in his skin », Progress in neurobiology, vol. 41, no 4, , p. 473–507 (ISSN 0301-0082, PMID 8210414) (Review)
- (en) G. Kreil, « Peptides containing a D-amino acid from frogs and molluscs », The Journal of biological chemistry, vol. 269, no 15, , p. 10967–10970 (ISSN 0021-9258, PMID 8157620, lire en ligne) (Review)
- (en) S. D. Heck, W. S. Faraci et al., « Posttranslational amino acid epimerization: enzyme-catalyzed isomerization of amino acid residues in peptide chains », PNAS, vol. 93, no 9, , p. 4036–4039 (ISSN 0027-8424, PMID 8633012, PMCID 39482)
- (en) R. Liardon et R. Jost, « Racemization of free and protein-bound amino acids in strong mineral acid », International journal of peptide and protein research, vol. 18, no 5, , p. 500–505 (ISSN 0367-8377, PMID 7341532)
- (en) H. Brückner, T. Westhauser et H. Godel, « Liquid chromatographic determination of D- and L-amino acids by derivatization with o-phthaldialdehyde and N-isobutyryl-L-cysteine. Applications with reference to the analysis of peptidic antibiotics, toxins, drugs and pharmaceutically used amino acids », Journal of chromatography. A, vol. 711, no 1, , p. 201–215 (ISSN 0021-9673, PMID 7496491).
- (en) A. Hashimoto, T. Nishikawa et al., « The presence of free D-serine in rat brain », FEBS letters, vol. 296, no 1, , p. 33–36 (ISSN 0014-5793, PMID 1730289)
- (en) N. W. Kleckner et R. Dingledine, « Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes », Science, vol. 241, no 4867, , p. 835–837 (ISSN 0036-8075, PMID 2841759)
- (en) H. Wolosker, S. Blackshaw et S. H. Snyder, « Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission », PNAS, vol. 96, no 23, , p. 13409–13414 (ISSN 0027-8424, PMID 10557334, PMCID 23961)
- (en) H. Wolosker, E. Dumin et al., « D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration », The FEBS journal, vol. 275, no 14, , p. 3514–3526 (ISSN 1742-464X, PMID 18564180, DOI 10.1111/j.1742-4658.2008.06515.x) (Review)
- (en) S. Sacchi, M. Bernasconi et al., « pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility », The Journal of biological chemistry, vol. 283, no 32, , p. 22244–22256 (ISSN 0021-9258, PMID 18544534, DOI 10.1074/jbc.M709153200)
- (en) H. J. Ryu, J. E. Kim et al., « Potential roles of D-serine and serine racemase in experimental temporal lobe epilepsy », Journal of neuroscience research, vol. 88, no 11, , p. 2469–2482 (ISSN 1097-4547, PMID 20623543, DOI 10.1002/jnr.22415)
- (en) S. A. Fuchs, R. Berger et T. J. de Koning, « D-serine: the right or wrong isoform? », Brain research, vol. 1401, , p. 104–117 (ISSN 1872-6240, PMID 21676380, DOI 10.1016/j.brainres.2011.05.039) (Review)
- (de) Julia Scharlau, « Untersuchungen zur Auswirkung von adoleszenter chronischer Cannabinoidbehandlung an einem Mausmodell der Schizophrenie » [PDF], sur Dissertation, Friedrich-Wilhelms-Universität Bonn, (consulté le ), p. 16 827 kB
- (en) E. Kartvelishvily, M. Shleper et al., « Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors », The Journal of biological chemistry, vol. 281, no 20, , p. 14151–14162 (ISSN 0021-9258, PMID 16551623, DOI 10.1074/jbc.M512927200)
- (en) K. Miya, R. Inoue et al., « Serine racemase is predominantly localized in neurons in mouse brain », The Journal of comparative neurology, vol. 510, no 6, , p. 641–654 (ISSN 1096-9861, PMID 18698599, DOI 10.1002/cne.21822)
- (en) L. Pollegioni et S. Sacchi, « Metabolism of the neuromodulator D-serine », Cellular and molecular life sciences, vol. 67, no 14, , p. 2387–2404 (ISSN 1420-9071, PMID 20195697, DOI 10.1007/s00018-010-0307-9) (Review)
- (en) J. T. Kantrowitz et D. C. Javitt, « 'N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? », Brain research bulletin, vol. 83, nos 3–4, , p. 108–121 (ISSN 1873-2747, PMID 20417696, PMCID 2941541, DOI 10.1016/j.brainresbull.2010.04.006) (Review)
- (en) J. T. Coyle, « Glutamate and schizophrenia: beyond the dopamine hypothesis », Cellular and molecular neurobiology, vol. 26, nos 4–6, , p. 365–384 (ISSN 0272-4340, PMID 16773445, DOI 10.1007/s10571-006-9062-8) (Review)
- (en) I. Chumakov, M. Blumenfeld et al., « Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia », PNAS, vol. 99, no 21, , p. 13675–13680 (ISSN 0027-8424, PMID 12364586, PMCID 129739, DOI 10.1073/pnas.182412499)
- (en) A. Corvin, K. A. McGhee et al., « Evidence for association and epistasis at the DAOA/G30 and D-amino acid oxidase loci in an Irish schizophrenia sample », American journal of medical genetics, vol. 144B, no 7, , p. 949–953 (ISSN 1552-4841, PMID 17492767, DOI 10.1002/ajmg.b.30452)
- (en) T. Ohnuma, N. Shibata et al., « Association analysis of glycine- and serine-related genes in a Japanese population of patients with schizophrenia », Progress in neuro-psychopharmacology & biological psychiatry, vol. 33, no 3, , p. 511–518 (ISSN 0278-5846, PMID 19223009, DOI 10.1016/j.pnpbp.2009.02.004)
- (en) K. Hashimoto, T. Fukushima et al., « Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia », Archives of general psychiatry, vol. 60, no 6, , p. 572–576 (ISSN 0003-990X, PMID 12796220, DOI 10.1001/archpsyc.60.6.572)
- (en) I. Bendikov, C. Nadri et al., « A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia », Schizophrenia research, vol. 90, nos 1–3, , p. 41–51 (ISSN 0920-9964, PMID 17156977, DOI 10.1016/j.schres.2006.10.010)
- (en) K. Hashimoto, G. Engberg et al., « Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients », Progress in neuro-psychopharmacology & biological psychiatry, vol. 29, no 5, , p. 767–769 (ISSN 0278-5846, PMID 15939521, DOI 10.1016/j.pnpbp.2005.04.023)
- (en) L. Verrall, M. Walker et al., « d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia », The European journal of neuroscience, vol. 26, no 6, , p. 1657–1669 (ISSN 0953-816X, PMID 17880399, PMCID 2121142, DOI 10.1111/j.1460-9568.2007.05769.x)
- (en) L. Verrall, P. W. Burnet et al., « The neurobiology of D-amino acid oxidase and its involvement in schizophrenia », Molecular psychiatry, vol. 15, no 2, , p. 122–137 (ISSN 1476-5578, PMID 19786963, PMCID 2811712, DOI 10.1038/mp.2009.99) (Review)
- (en) R. Kapoor, K. S. Lim et al., « Preliminary evidence for a link between schizophrenia and NMDA-glycine site receptor ligand metabolic enzymes, d-amino acid oxidase (DAAO) and kynurenine aminotransferase-1 (KAT-1) », Brain research, vol. 1106, no 1, , p. 205–210 (ISSN 0006-8993, PMID 16828464, DOI 10.1016/j.brainres.2006.05.082)
- (en) C. Madeira, M. E. Freitas et al., « Increased brain D-amino acid oxidase (DAAO) activity in schizophrenia », Schizophrenia research, vol. 101, nos 1–3, , p. 76–83 (ISSN 0920-9964, PMID 18378121, DOI 10.1016/j.schres.2008.02.002)
- (en) J. T. Coyle, G. Tsai et D. C. Goff, « Ionotropic glutamate receptors as therapeutic targets in schizophrenia », Current drug targets, vol. 1, no 2, , p. 183–189 (ISSN 1568-007X, PMID 12769626) (Review)
- (en) G. Tsai, P. Yang et al., « D-serine added to antipsychotics for the treatment of schizophrenia », Biological psychiatry, vol. 44, no 11, , p. 1081–1089 (ISSN 0006-3223, PMID 9836012)
- (en) H. J. Tuominen, J. Tiihonen et K. Wahlbeck, « Glutamatergic drugs for schizophrenia », Cochrane database of systematic reviews (Online), no 2, , p. CD003730 (ISSN 1469-493X, PMID 16625590, DOI 10.1002/14651858.CD003730.pub2) (Review)
- (en) D. V. Ferraris et T. Tsukamoto, « Recent advances in the discovery of D-amino acid oxidase inhibitors and their therapeutic utility in schizophrenia », Current pharmaceutical design, vol. 17, no 2, , p. 103–111 (ISSN 1873-4286, PMID 21361869) (Review)
- (en) C. A. Strick, C. Li et al., « Modulation of NMDA receptor function by inhibition of D-amino acid oxidase in rodent brain », Neuropharmacology, vol. 61, nos 5–6, , p. 1001–1015 (ISSN 1873-7064, PMID 21763704, DOI 10.1016/j.neuropharm.2011.06.029).
- (en) T. Sparey, P. Abeywickrema et al., « The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors », Bioorganic & medicinal chemistry letters, vol. 18, no 11, , p. 3386–3391 (ISSN 1464-3405, PMID 18455394, DOI 10.1016/j.bmcl.2008.04.020)
- (en) J.F. Berry, D.V. Ferraris, B. Duvall et al., « Synthesis and SAR of 1-hydroxy-1H-benzo[d]imidazol-2(3H)-ones as Inhibitors of D-Amino Acid Oxidase », ACS Med Chem Lett., vol. 3, no 10, , p. 839–843 (PMID 23243487, DOI 10.1021/ml300212a)
- (en) S. Sacchi, E. Rosini, L. Pollegioni et G. Molla, « D-Amino Acid Oxidase Inhibitors as a Novel Class of Drugs for Schizophrenia Therapy », Curr. Pharm. Des., (PMID 23116391)
- (en) P. Paul et J. de Belleroche, « The role of D-amino acids in amyotrophic lateral sclerosis pathogenesis: a review », Amino Acids, vol. 43, no 5, , p. 1823–1831 (PMID 22890612, DOI 10.1007/s00726-012-1385-9)
- (en) J. Sasabe, T. Chiba, M. Yamada et al., « D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis », EMBO J., vol. 26, no 18, , p. 4149–4159 (PMID 17762863, PMCID 2230675, DOI 10.1038/sj.emboj.7601840)
- (en) D. S. Dunlop, A. Neidle et al., « The presence of free D-aspartic acid in rodents and man », Biochemical and biophysical research communications, vol. 141, no 1, , p. 27–32 (ISSN 0006-291X, PMID 3801000)
- (en) H. Wolosker, A. D'Aniello et S. H. Snyder, « D-aspartate disposition in neuronal and endocrine tissues: ontogeny, biosynthesis and release », Neuroscience, vol. 100, no 1, , p. 183–189 (ISSN 0306-4522, PMID 10996468).
- (en) M. Katane et H. Homma, « D-aspartate oxidase: the sole catabolic enzyme acting on free D-aspartate in mammals », Chemistry & biodiversity, vol. 7, no 6, , p. 1435–1449 (ISSN 1612-1880, PMID 20564562, DOI 10.1002/cbdv.200900250) (Review)
- (en) H. Ohide, Y. Miyoshi et al., « D-Amino acid metabolism in mammals: biosynthesis, degradation and analytical aspects of the metabolic study », Journal of chromatography. B, vol. 879, no 29, , p. 3162–3168 (ISSN 1873-376X, PMID 21757409, DOI 10.1016/j.jchromb.2011.06.028) (Review)
- (en) P. M. Kim, X. Duan et al., « Aspartate racemase, generating neuronal D-aspartate, regulates adult neurogenesis », PNAS, vol. 107, no 7, , p. 3175–3179 (ISSN 1091-6490, PMID 20133766, PMCID 2840285, DOI 10.1073/pnas.0914706107)
- (en) Y. Mori, K. Aki et al., « UV B-irradiation enhances the racemization and isomerizaiton of aspartyl residues and production of N?-carboxymethyl lysine (CML) in keratin of skin », Journal of chromatography B, vol. 879, no 29, , p. 3303–3309 (ISSN 1873-376X, PMID 21636332, DOI 10.1016/j.jchromb.2011.05.010)
- (en) N. Fujii, Y. Kaji et al., « Collapse of homochirality of amino acids in proteins from various tissues during aging », Chemistry & biodiversity, vol. 7, no 6, , p. 1389–1397 (ISSN 1612-1880, PMID 20564552, DOI 10.1002/cbdv.200900337) (Review)
- (en) N. Fujii, « D-amino acid in elderly tissues », Biological & pharmaceutical bulletin, vol. 28, no 9, , p. 1585–1589 (ISSN 0918-6158, PMID 16141520) (Review)
- (en) Graham C. Barrett et Donald Trevor Elmore, Amino Acids and Peptides, Cambridge University Press, (ISBN 0-521-46292-4, lire en ligne), p. 15–16
- (en) S. Ritz-Timme et M. J. Collins, « Racemization of aspartic acid in human proteins », Ageing research reviews, vol. 1, no 1, , p. 43–59 (ISSN 1568-1637, PMID 12039448) (Review)
- (en) H. Mori, K. Ishii et al., « Racemization: its biological significance on neuropathogenesis of Alzheimer's disease », The Tohoku journal of experimental medicine, vol. 174, no 3, , p. 251–262 (ISSN 0040-8727, PMID 7761990)
- (en) R. Shapira, G. E. Austin et S. S. Mirra, « Neuritic plaque amyloid in Alzheimer's disease is highly racemized », Journal of neurochemistry, vol. 50, no 1, , p. 69–74 (ISSN 0022-3042, PMID 3121789).
- (en) A. E. Roher, J. D. Lowenson et al., « Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease », The Journal of biological chemistry, vol. 268, no 5, , p. 3072–3083 (ISSN 0021-9258, PMID 8428986, lire en ligne)
- (en) T. Tomiyama, S. Asano et al., « Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid beta protein analogues », The Journal of biological chemistry, vol. 269, no 14, , p. 10205–10208 (ISSN 0021-9258, PMID 8144598)
- (en) Patrick R. Hof et Charles V. Mobbs, Handbook of the Neuroscience of Aging, Academic Press, (ISBN 0-123-74898-4, lire en ligne), p. 41
- (en) M. L. Moro, M. J. Collins et E. Cappellini, « Alzheimer's disease and amyloid beta-peptide deposition in the brain: a matter of 'aging'? », Biochemical Society transactions, vol. 38, no 2, , p. 539–544 (ISSN 1470-8752, PMID 20298218, DOI 10.1042/BST0380539) (Review)
- (de) Günter Westphal, Gerhard Gerber et Bodo Lipke, Proteine – Nutritive und funktionelle Eigenschaften, Springer, (ISBN 3-540-00232-4, lire en ligne), p. 125
- (de) Gerhard G. Habermehl, Peter E. Hammann et al., Naturstoffchemie, Springer, , 3e éd. (ISBN 3-540-73732-4, lire en ligne), p. 252
- Belitz2001, p. 33.
- (en) N. J. Benevenga et R. D. Steele, « Adverse effects of excessive consumption of amino acids », Annual review of nutrition, vol. 4, , p. 157–181 (ISSN 0199-9885, PMID 6235826, DOI 10.1146/annurev.nu.04.070184.001105) (Review)
- (en) J. Zagon, L.-I. Dehne et K.-W. Bögl, « D-Amino acids in organisms and food », Nutr Res., vol. 14, , p. 445–463
- (de) Werner Baltes et Reinhard Matissek, Lebensmittelchemie, Springer, , 7e éd. (ISBN 3-642-16538-9, lire en ligne), p. 164
- (en) W. Leuchtenberger, K. Huthmacher et K. Drauz, « Biotechnological production of amino acids and derivatives: current status and prospects », Applied microbiology and biotechnology, vol. 69, no 1, , p. 1–8 (ISSN 0175-7598, PMID 16195792, DOI 10.1007/s00253-005-0155-y) (Review)
- (en) F. H. Leibach et V. Ganapathy, « Peptide transporters in the intestine and the kidney », Annual review of nutrition, vol. 16, , p. 99–119 (ISSN 0199-9885, PMID 8839921, DOI 10.1146/annurev.nu.16.070196.000531) (Review)
- (en) M. Friedman, « Origin, microbiology, nutrition, and pharmacology of D-amino acids », Chemistry & biodiversity, vol. 7, no 6, , p. 1491–1530 (ISSN 1612-1880, PMID 20564567, DOI 10.1002/cbdv.200900225) (Review)
- (en) W. Heine, K. Wutzke et U. Drescher, « D-amino acid utilization in infants measured with the 15N-tracer technique », Clinical nutrition (Edinburgh, Scotland), vol. 2, no 1, , p. 31–35 (ISSN 0261-5614, PMID 16829405)
- (en) M. Friedman et R. Liardon, « Racemization kinetics of amino acid residues in alkali-treated soybean proteins », J Agric Food Chem., vol. 33, , p. 666–672.
- (en) J. C. Crawhall et D. F. Elliott, « A note on the racemization of serine », The Biochemical journal, vol. 48, no 2, , p. 237–238 (ISSN 1470-8728, PMID 14820833, PMCID 1275510).
- (en) M. Lüpke et H. Brückner, « Gas chromatographic evaluation of amino acid epimerisation in the course of gelatin manufacturing and processing », Zeitschrift für Lebensmitteluntersuchung und -Forschung A, vol. 206, no 5, , p. 323-328 (DOI 10.1007/s002170050266)
- (de) Ernährungsbericht 1996, Deutsche Gesellschaft für Ernährung, (ISBN 3-921606-33-0), p. 149
- (de) Johannes Friedrich Diehl, Chemie in Lebensmitteln, John Wiley & Sons, (ISBN 3-527-66084-4, lire en ligne), p. 95
- (en) H. Lapierre, G. Holtrop et al., « Is D-methionine bioavailable to the dairy cow? », Journal of dairy science, vol. 95, no 1, , p. 353–362 (ISSN 1525-3198, PMID 22192214, DOI 10.3168/jds.2011-4553)
- (en) W. H. Fishman et C. Artom, « Serine injury », J Biol Chem., vol. 145, , p. 345–346
- (en) R. P. Morehead, C. Artom, et W. H. Fishman, « The nephrotoxic action of serine », Proceedings. American Federation for Clinical Research, vol. 2, , p. 81 (PMID 20275626)
- (en) F. A. Carone et C. E. Ganote, « D-serine nephrotoxicity. The nature of proteinuria, glucosuria, and aminoaciduria in acute tubular necrosis », Archives of pathology, vol. 99, no 12, , p. 658–662 (ISSN 0363-0153, PMID 1203037).
- (en) R. E. Williams et E. A. Lock, « D-serine-induced nephrotoxicity: possible interaction with tyrosine metabolism », Toxicology, vol. 201, nos 1–3, , p. 231–238 (ISSN 0300-483X, PMID 15297036, DOI 10.1016/j.tox.2004.05.001).
- (en) D. R. Peterson et F. A. Carone, « Renal regeneration following d-serine induced acute tubular necrosis », The Anatomical record, vol. 193, no 3, , p. 383–388 (ISSN 0003-276X, PMID 426302, DOI 10.1002/ar.1091930305)
- (en) C. E. Ganote, D. R. Peterson et F. A. Carone, « The nature of D-serine–induced nephrotoxicity », The American Journal of Pathology, vol. 77, no 2, , p. 269–282 (ISSN 0002-9440, PMID 4447130, PMCID 1910915)
- (en) M. S. Pilone et L. Pollegioni, « D-amino acid oxidase as an industrial biocatalyst », Biocatalysis and Biotransformation, vol. 20, no 3, , p. 145–159 (DOI 10.1080/10242420290020679).
- (en) A. W. Krug, K. Völker et al., « Why is D-serine nephrotoxic and alpha-aminoisobutyric acid protective? », American journal of physiology. Renal physiology, vol. 293, no 1, , F382–F390 (ISSN 1931-857X, PMID 17429029, DOI 10.1152/ajprenal.00441.2006, lire en ligne)
- (en) G. Lubec, C. Wolf et B. Bartosch, « Aminoacid isomerisation and microwave exposure », Lancet, vol. 332, no 8676, , p. 1392–1393 (ISSN 0140-6736, PMID 2574327)
- (en) Karl S. Kruszelnicki, « Microwaves damage food », sur ABC abcscience, (consulté le )
- (en) W. Segal, « Microwave heating of milk », Lancet, vol. 335, no 8687, , p. 470 (ISSN 0140-6736, PMID 1968186)
- (de) « Die Wirkung von mit Mikrowellen bestrahltem Wasser auf Pflanzen » (consulté le ) Retiré le 24/8/2012
- (de) « Mikrowellennahrung macht dick und erzeugt Krebs », (consulté le ) Retiré le 28/9/2012
- (de) « Gefährliche Mikrowellen », (consulté le ) Retiré le 28/9/2012
- (de) P. Fritz, L. I. Dehne, J. Zagon et K. W. Bögl, « Zur Frage der Aminosäureisomerisierung im Mikrowellenfeld: Ergebnisse eines Modellversuches mit Standardlösungen », Zeitschrift für Ernährungswissenschaft, vol. 31, no 3, , p. 219–224.
- (en) L. I. Dehne et P. Fritz, « Isomerisierung von Aminosäuren durch Mikrowellenerhitzung? », Bundesgesundheitsblatt, no 35, , p. 463–464.
- (en) P. Sieber, P. Eberhardt et P. U. Gallmann, « Heat treatment of milk in domestic microwave ovens », Int Dairy Journal, vol. 6, , p. 231–246 (lire en ligne).
- (en) J. Zagon, L.-I. Dehne et K.-W. Bögl, « Isomerisierung von Aminosäuren in Lebensmitteln. Teil I: Mechanismen und Verbreitung der Aminosäurenisomerisierung in Organismen und Lebensmitteln. », Ernährungs-Umschau, vol. 38, , p. 275–278, 324–328
- (en) A. Cherkin, J. L. Davis et M. W. Garman, « D-Proline: stereospecificity and sodium chloride dependence of lethal convulsant activity in the chick », Pharmacology, biochemistry, and behavior, vol. 8, no 5, , p. 623–625 (ISSN 0091-3057, PMID 674269)
- (en) A. Schieber, H. Brückner et al., « Evaluation of D-amino acid levels in rat by gas chromatography-selected ion monitoring mass spectrometry: no evidence for subacute toxicity of orally fed D-proline and D-aspartic acid », Journal of chromatography, vol. 691, no 1, , p. 1–12 (ISSN 1387-2273, PMID 9140753)
- (en) S. N. Ali, G. O'Toole et M. Tyler, « Milk bottle burns », The Journal of burn care & rehabilitation, vol. 25, no 5, , p. 461–462 (ISSN 0273-8481, PMID 15353942)
- (en) I. A. Jaffe, K. Altman et P. Merryman, « The antipyridoxine effect of penicillamine in man », The Journal of clinical investigation, vol. 43, , p. 1869–1873 (ISSN 0021-9738, PMID 14236210, PMCID 289631, DOI 10.1172/JCI105060).
- (en) T. N. Schumacher, L. M. Mayr et al., « Identification of D-peptide ligands through mirror-image phage display », Science, vol. 271, no 5257, , p. 1854–1857 (ISSN 0036-8075, PMID 8596952)
- (de) Katja Wiesehan, « Identifizierung und Charakterisierung eines spezifischen Liganden für das Alzheimer Amyloid-β-Peptid (Aβ) » [PDF], Dissertation, sur Universität Bayreuth, (consulté le ), p. 21 11,1 MB
- (en) J. M. Manning et S. Moore, « Determination of D- and L-amino acids by ion exchange chromatography as L-D and L-L dipeptides », The Journal of biological chemistry, vol. 243, no 21, , p. 5591–5597 (ISSN 0021-9258, PMID 5699053)
- (en) K. Soda, « Microdetermination of D-amino acids and D-amino acid oxidase activity with 3,methyl-2-benzothiazolone hydrazone hydrochloride », Analytical biochemistry, vol. 25, no 1, , p. 228–235 (ISSN 0003-2697, PMID 4387536)
- (en) D. L. Kirschner et T. K. Green, « Separation and sensitive detection of D-amino acids in biological matrices », Journal of separation science, vol. 32, no 13, , p. 2305–2318 (ISSN 1615-9314, PMID 19569111, DOI 10.1002/jssc.200900101) (Review)
- (en) K. Hamase, A. Morikawa et al., « Analysis of small amounts of D-amino acids and the study of their physiological functions in mammals », Analytical sciences, vol. 25, no 8, , p. 961–968 (ISSN 1348-2246, PMID 19667471) (Review)
- (de) Clarissa Dorothee Marzini, « Grenzen der quantitativen Enantiomeranalytik von a-Aminosäuren » [PDF], sur Dissertation, Eberhard-Karls-Universität Tübingen, (consulté le ), p. 20–21 1,7 MB
- (en) N. Nimura et T. Kinoshita, « O-Phthaldialdehyde N-acetyl-L-cysteine as a chiral derivatization reagent for liquid chromatographic optical resolution of amino acid enantiomers and ist application to conventional amino acid analysis », Journal of Chromatography A, vol. 352, , p. 169–177 (DOI 10.1016/S0021-9673(01)83377-X)
- (de) Birgit Geueke (Dissertation (PDF; 9,6 MB)), « Neue Aminosäureoxidasen aus Rhodococcus opacus und Arthrobacter protophormiae: Untersuchungen zur biochemischen Charakterisierung, Klonierung und Expression », sur Heinrich-Heine-Universität Düsseldorf, (consulté le ), p. 1
- (en) E. Takahashi, M. Furui et al., « D-lysine production from L-lysine by successive chemical racemization and microbial asymmetric degradation », Applied Microbiology and Biotechnology, vol. 47, no 4, , p. 347–351 (ISSN 0175-7598, PMID 9163947).
- (en) K. Isobe, H. Tamauchi et al., « A Simple Enzymatic Method for Production of a Wide Variety of D-Amino Acids Using L-Amino Acid Oxidase from Rhodococcus sp. AIU Z-35-1 », Enzyme research, vol. 2010, , p. 567210 (ISSN 2090-0414, PMID 21048866, PMCID 2962901, DOI 10.4061/2010/567210)
- (de) Kerstin Ragnitz, « Immobilisierung und Stabilisierung der Hydantoinase und L-N-Carbamoylase aus Arthrobacter aurescens DSM 3747 » [PDF], sur Dissertation, Universität Stuttgart, (consulté le ), p. 4 997 kB
- (en) « Global D-Amino Acids Market to Reach $3.7 Billion by 2017, According to a New Report by Global Industry Analysts, Inc. », sur prweb.com, (consulté le )
- (en) S. Martínez-Rodríguez, A. I. Martínez-Gómez et al., « Natural occurrence and industrial applications of D-amino acids: an overview », Chemistry & biodiversity, vol. 7, no 6, , p. 1531–1548 (ISSN 1612-1880, PMID 20564568, DOI 10.1002/cbdv.200900245) (Review)
- (en) Markus Werner, « Klonierung der D-Carbamoylase aus Arthrobacter crystallopoietes DSM 20117 », Dissertation, sur Universität Stuttgart, (consulté le )
- (de) B. Kutscher et M. Bernd, « Chemie und Molekularbiologie bei der Suche nach neuen LHRH-Antagonisten », Angew Chem., vol. 109, no 20, , p. 2240–2254 (DOI 10.1002/ange.19971092005).
- (en) T. Beckers, M. Bernd et al., « Structure-function studies of linear and cyclized peptide antagonists of the GnRH receptor », Biochemical and biophysical research communications, vol. 289, no 3, , p. 653–663 (ISSN 0006-291X, PMID 11726197, DOI 10.1006/bbrc.2001.5939)
- (en) Alaa A.-M. Abdel-Aziz, Yousif A. Asiri et al., « Tadalafil », dans Harry G. Brittain, Profiles of Drug Substances, Excipients and Related Methodology, Academic Press, , PDF (ISBN 0-123-87702-4, lire en ligne), p. 287–329 4,4 MB
- (en) E. Fischer, B. Heller et al., « Therapy of depression by phenylalanine : Preliminary note », Arzneimittel-Forschung, vol. 25, no 1, , p. 132 (ISSN 0004-4172, PMID 1173765)
- (en) S. Ehrenpreis, « D-phenylalanine and other enkephalinase inhibitors as pharmacological agents: implications for some important therapeutic application », Substance and alcohol actions/misuse, vol. 3, no 4, , p. 231–239 (ISSN 0191-8877, PMID 6301083)
- (en) Mitchell Bebel Stargrove, Jonathan Treasure et Dwight L. McKee, Herb, Nutrient, and Drug Interactions, Elsevier Health Sciences, (ISBN 0-323-02964-7, lire en ligne), p. 682
- (en) « Phenylalanine », sur University of Maryland Medical Center (consulté le ) Retiré le 21/9/2012
- (en) D. L. Williams, « A veterinary approach to the European honey bee (Apis mellifera) », Veterinary journal, vol. 160, no 1, , p. 61–73 (ISSN 1090-0233, PMID 10950136, DOI 10.1053/tvjl.2000.0474) (Review)
 Portail de la chimie
Portail de la chimie  Portail de la biochimie
Portail de la biochimie