Composé organoboré
Les composés organoborés sont une classe de composés organiques comportant au moins une liaison entre un atome de carbone et un atome de bore. Le terme « organoborane » est parfois utilisé comme synonyme de composé organoboré, mais il est aussi utilisé dans un sens plus restreint pour désigner les seuls dérives alkylés ou arylés du borane (BH3), comme les trialkylboranes (BR3). Parmi les autres grandes familles des composés organoborés, on compte les acides boroniques et les esters boroniques
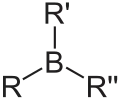 Organoborane (au sens restreint)
Organoborane (au sens restreint)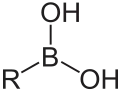 Acide boronique
Acide boronique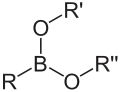 Ester boronique
Ester boronique
Les composés organoborés sont d'importants réactifs en chimie organique permettant de nombreuses transformations chimiques dont la plus importante est l'hydroboration.
Liaison carbone–bore

La liaison carbone–bore (C-B) est faiblement polaire du fait de la faible différence d'électronégativité entre le carbone (2,55) et le bore (2,04). De ce fait, les alkylboranes sont généralement stables, mais facilement oxydables.
En partie du fait de sa plus faible électronégativité, le bore forme souvent des composés à déficit d'électrons (en) tels que les triorganoboranes. Les groupes vinyle et aryle tendent eux à donner leurs électrons, et rendent ainsi le bore moins électrophile, donnant alors à la liaison C-B un caractère de liaison double. Les dérivés organiques du diborane sont classés en chimie organique comme des électrophiles forts car le bore y est incapable d'obtenir un octet d'électrons.
Classes de composés
Organoboranes et hydrures
L'une des classes de composés organoborés la plus étudiée est celle des composés de formule BRnH3−n. Ces composés sont utilisés comme catalyseurs, réactifs ou intermédiaires réactionnels. Les dérivés trialkyle et triaryle présentent une géométrie plane trigonale autour du centre de bore, et sont typiquement des acides de Lewis faibles. À l'exception de certains dérivés très volumineux, les hydrures (BRnH3−n avec n = 1 or 2) existent sous la forme de dimères, comme le borane lui-même. Les dérivés trisubstitués, comme le triéthylborane sont eux monomériques[2].
Acides et esters boriniques et boroniques (BRn(OR)3-n)
Les composés de type type BRn(OH)3-n sont appelés acides boriniques (n = 2) et acides boroniques (n = 1), et ceux de type BRn(OR)3-n esters boriniques et boroniques. Les cas pour n = 0 correspondent respectivement à l'acide borique et aux borates qui ne sont pas considérés comme des composés organoborés puisque ne comportant pas de liaison carbone–bore. Le borate de triméthyle, B(OCH3)3, est utilisé comme précurseur des esters boroniques pour le couplage de Suzuki.
Clusters de bore
Le bore est réputé pour former des clusters, par exemple le dodécaborate [B12H12]2−. De nombreux dérivés organiques sont connus pour former de tels clusters. Une exemple est [B12(CH3)12]2− et son dérivé radical [B12(CH3)12]−[3]. Les composés cluster apparentés comprenant des atomes de carbone sont appelés carboranes. Le plus connu est l'orthocarborane, de formule C2B10H12. Bien qu'ils aient peu d'applications commerciales, les carboranes ont beaucoup attiré d'attention en raison de leur structure inhabituelle. Leurs dérivés anioniques, les dicarbollures, par exemple [C2B9H11]2−, sont des ligands qui se comportent comme le cyclopentadiénure.
Composés aromatiques borasubstitués
Il existe des composés aromatiques dérivés d'hydrocarbures aromatiques où l'un groupe CH est remplacé par un atome de bore. C'est par exemple le cas du borabenzène (en) (C5H5B). Ce type de composés est toujours isolé sous la forme d'adduits, par exemple C5H5B-pyridine. Le borole, un analogue structurel du pyrrole ou de l'azole, n'a jamais été isolé, mais on en connaît des dérivés substitués. La borépine (hétérocycle à sept) elle été isolée et, contrairement à l'azépine ou l'oxépine, est aromatique.
Boryles
Les anions boryles ont pour formule R2B−. Les composés boryles anioniques nucléophiles se sont longtemps montrés insaisissables, mais une étude de 2006 a décrit un composé de boryllithium, qui réagit comme un nucléophile[4],[5]. Les composés organométalliques avec des liaisons métal-bore (c'est-à-dire M–BR2) sont connus sous le nom de complexes boryle. Les ligands associés sont appelés borylènes (M–B(R)–M).
L'absence de composés de boyle de lithium est notable car les sels de lithium sont courants chez les autres éléments de la seconde période, par exemple le fluorure de lithium, l'hydroxyde de lithium, l'amidure de lithium ou le méthyllithium. Cette absence souligne la très faible électronégativité du bore. Les réactions des bases avec les hydrures de bore (R2BH) n'a pas pour résultat la déprotonation en anion boryle R2B−, mais la formation de l'anion boryle R2B−H(base)+, qui possède un octet complet[6]. Au lieu de cela, le composé de bromyle est préparé par hétérolyse réductrice d'une liaison bore-brome par le lithium métallique. Ce composé boryllithium est tres similaire et isoélectronique aux carbènes N-hétérocycliques. Il bénéficie de la stabilisation aromatique (6 électron en comptant les doublets libres de l'azote et l'orbitale p vide du bore – structure A) et de la stabilité cinétique des encombrants groupes 2,6-diisopropylphényle. La cristallographie aux rayons X permet de confirmer une hybridation sp2 du bore, et le fait qu'il réagisse par addition nucléophile avec le benzaldéhyde est une preuve supplémentaire qu'il adopte ben la structure proposée.
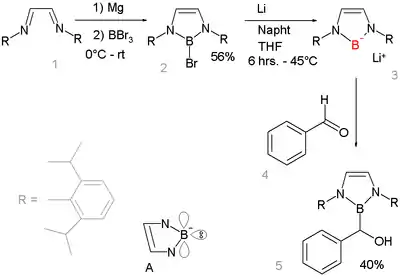
Alkylidèneboranes
Les alkylidèneboranes, les composés de type RB=CRR avec une liaison double carbone–bore sont rares. Le composé parent de cette sous-classe est HB=CH2, qui peut être détecté à basses températures. Un dérivé assez stable est CH3B=C(SiMe3)2, mais il tendance à se cyclodimériser[7].
Adduits NHC du bore
Les carbènes N-hétérocycliques (NHC) et les boranes forment des adduits stables, les NHC boranes[8]. Les adduits de triéthylborane peuvent être synthétisés directement à partir du sel d'imidazolium et du triéthylborohydrure de lithium. Les membres de cette classe de composés sont étudiés pour être utilisés comme réactifs ou catalyseurs.
Diborènes
Les composés contenant une liaison double bore–bore sont rares. En 2007, le premier diborène (RHB=BHR) neutre a été présenté par Gregory Robinson de l'université de Géorgie[9],[10]. Chaque atome de bore a un proton attaché à lui, et est coordonné à un carbène N-hétérocyclique (NHC). La structure parente avec des ligands carbène supplémentaire est le diborane(2)[11],[12].
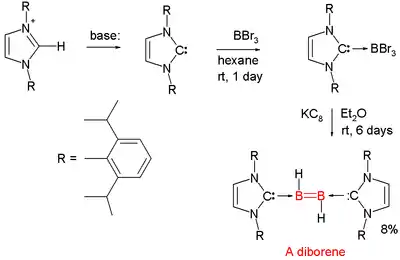
Un diboryne a été rapporté, basé sur une chimie similaire.
Synthèse
À partir de réactifs de Grignard
Des organoboranes simples tels que le triéthylborane ou le tris(pentafluorophényl)borane peuvent être préparés à partir du trifluoroborane et du réactif de Grignard d'éthyle ou de pentafluorophényle correspondant. Les borates (R4B−) sont eux produits par addition d'équivalents R− (RMgX, RLi, etc.) à R3B.
À partir d'alcènes
De alcènes peuvent être insérés dans des liaisons B-H de boranes dans un procédé appelé hydroboration. L'hydroboration des alcènes ou des alcynes par le borane (BH3) ou ses dérivés ne convertit qu'environ 33% des réactifs initiaux, le reste étant inclus après oxydation ou protonolyse dans des sous-produits borés. Un réactif organoboré couramment utilisé en synthèse est le 9-BBN[13]. L'hydroboration est stéréospécifique syn, et donne le produit anti-Markovnikov.
Par borylation
La réaction de borylation de C-H catalysée par les métaux est une réaction organique catalysée les métaux de transition qui produit de composés organoborés par fonctionnalisation de liaisons C–H aliphatiques ou aromatiques. Un réactif courant pour ce genre de réactions est le bis(pinacolato)diborane (en).
Réactivité
La liaison carbone–bore est légèrement polarisée vers le carbone, ce qui le rend nucléophile. Cette propriété est exploitée pour transférer l'un des groupes R vers un centre électrophile de façon inter- ou, plus souvent, intramoléculaire[14]. Dans ce dernier cas, le groupe nuclophile R est capable de subir une migration 1,2 vers le carbone électrophile attaché au bore. Le borane résultant peut ensuite être ensuite oxydé ou être sujet d'une protonolyse pour donner divers composés organiques :
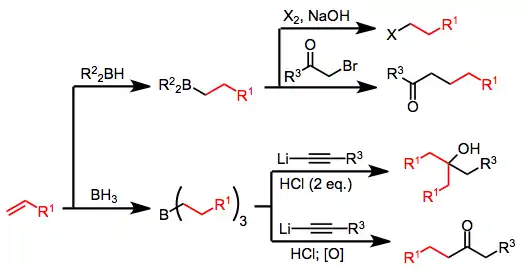
Hydroboration-oxydation
En synthèse organique, la réaction d'hydroboration est poussée plus loin pour générer d'autres groupes fonctionnels à la place du groupe du bore. La réaction d'hydroboration-oxydation (en) offre une voie vers les alcools par oxydation du borane avec le peroxyde d'hydrogène, ou vers les composés carbonylés avec un oxydant plus fort comme le trioxyde de chrome.
Réarrangements
Le monoxyde de carbone réagit avec les trialkylboranes. Il en suit un réarrangement 1,2 dans lequel un substituant alkyle migre du bore au carbone du groupe carbonyle. Il est ainsi possible des alcools primaires homologues par traitement des organoboranes par le monoxyde de carbone et un hydrure[15] :

Allylboration
L'allylboration asymétrique est une autre façon pratique de former des liaisons carbone-carbone grâce aux organoborés[16]. On peut citer par exemple la synthèse de Nicolaou des épothilones[17] où une allylboration asymétrique (utilisant allylborane dérivé de l'α-pinène) est utilisée en conjonction avec une protection par le TBS et une ozonolyse. Cela résulte au général en une homologation ajoutant deux carbones, produisant la séquence d'acétogénine recherchée.
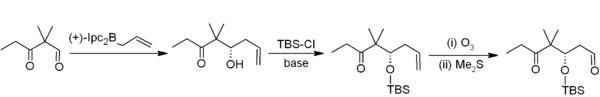
Réducteur
Les hydrure de borane tel que le 9-BBN ou le L-Selectride (tri-sec-butylborohydrure de lithium) sont utilisés comme réducteurs. Le catalyseur CBS qui contient du bore est lui utilisé comme catalyseur asymétrique pour la réduction des carbonyles.
Borates
Les trialkylboranes (BR3) peuvent être oxydés en leurs borates correpondants (B(OR)3). Cette oxydation peut aussi servir en analyse chimique à déterminer le nombre de liaisons carbone-bore que contient un composé à analyser ; le composé est ainsi mis à réagir avec l'oxyde de triméthylamine (Me3NO), la triméthylamine (Me3N) formée étant ensuite titrée.
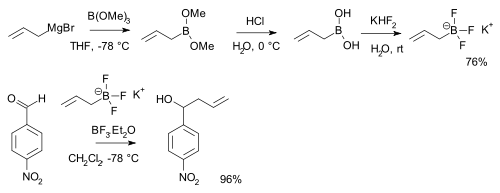
Les borates peuvent être convertis en acides boroniques (RB(OH)2) par réaction avec un réactif de Grignard puis hydrolyse. Ces acides peuvent ensuite réagir avec le bifluorure de potassium (K[HF2]) pour former des sels de trifluoroborate (K[RBF3])[18], des précurseurs des nucléophiles difluorures d'alkyle et d'aryle, ArBF2[19]. Ces sels sont plus stables que les acides boroniques eux-mêmes, et sont utilisés par exemple pour l'alkylation de certains aldéhydes[20],[note 1] :
Réaction de Suzuki et réactions connexes
Les composés organoborés se prêtent également aux réactions de transmétallation, en particulier avec les composés d'organopalladium. Ce type de réaction est illustré par la réaction de Suzuki, qui implique le couplage d'un acide aryl- ou vinyl-boronique avec un halogénure d'aryle ou de vinyle, catalysé par un complexe de palladium(0)[21].
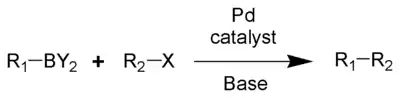
Cette réaction est une méthode importante dans la création de liaisons carbone-carbone.
Autres utilisations
Le triéthylborane (TEB) a été utilisé pour allumer le combustible JP-7 du moteur à cycle variable Pratt & Whitney J58 qui équipe le Lockheed SR-71 Blackbird.
Notes
- La synthèse montrée ici commence avec la réaction du bromure d'allylmagnésium avec le borate de triméthyle, suivie par l'hydrolyse de l'ester boronique en acide boronique par l'acide chlorhydrique. L'aldéhyde alkylé est le p-nitrobenzaldéhyde.
Références
- (en) Cet article est partiellement ou en totalité issu de l’article de Wikipédia en anglais intitulé « Organoboron chemistry » (voir la liste des auteurs).
- Ruth A. Bartlett, H. V. Rasika Dias, Marilyn M. Olmstead, Philip P. Power et Kenneth J. Weese, « Synthesis of the monomeric HBtrip2 (Trip - 2,4,6-iso-Pr3C6H2) and the x-ray crystal structures of [HBMes2]2 (Mes = 2,4,6,-Me3C6H2) and HBtrip2 », Organometallics, vol. 9, , p. 146–150 (DOI 10.1021/om00115a023)
- (en) Brown, H. C., Organic Syntheses via Boranes, New York, John Wiley & Sons, (ISBN 0-471-11280-1)
- Grimes, R. N., Carboranes, 3rd edition, New York, Academic Press, , 1058 p. (ISBN 978-0-12-801905-4, lire en ligne)
- Segawa Yasutomo, Yamashita Makoto, Nozaki Kyoko, « Boryllithium: Isolation, Characterization, and Reactivity as a Boryl Anion », Science, vol. 314, no 5796, , p. 113–115 (PMID 17023656, DOI 10.1126/science.1131914, Bibcode 2006Sci...314..113S)
- Bethany Halford Boron Attacks Electropositive element pressed into action as nucleophilic boryllithium Chemical & Engineering News 2006; Volume 84(41): 11 Link
- Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine. Dennis G. Hall (ISBN 3-527-30991-8)
- Paetzold Peter, Englert Ulli, Finger Rudolf, Schmitz Thomas, Tapper Alexander, Ziembinski Ralf, « Reactions at the Boron-Carbon Double Bond of Methyl(methylidene)boranes », Z. Anorg. Allg. Chem., vol. 630, no 4, , p. 508–518 (DOI 10.1002/zaac.200300396)
- Curran D. P., Solovyev A., Makhlouf Brahmi M., Fensterbank L., Malacria M., Lacôte E., « Synthesis and Reactions of N-Heterocyclic Carbene Boranes », Angewandte Chemie International Edition, vol. 50, no 44, , p. 10294–10317 (PMID 21898724, DOI 10.1002/anie.201102717)
- Yuzhong Wang, Brandon Quillian, Pingrong Wei, Chaitanya S. Wannere, Yaoming Xie, R. Bruce King, Henry F. Schaefer, III, Paul v. R. Schleyer, and Gregory H. Robinson, « A Stable Neutral Diborene Containing a B=B Double Bond », J. Am. Chem. Soc., vol. 129, no 41, , p. 12412–12413 (PMID 17887683, DOI 10.1021/ja075932i, lire en ligne)
- Neutral Diborene Is A First Ron Dagani Chemical & Engineering News October 1, 2007 Volume 85, Number 40 p. 10
- (en) Holger Braunschweig et Rian D. Dewhurst, « Single, Double, Triple Bonds and Chains: The Formation of Electron-Precise B-B Bonds », Angewandte Chemie International Edition, vol. 52, no 13, , p. 3574–3583 (ISSN 1521-3773, PMID 23362015, DOI 10.1002/anie.201208189)
- (en) Merle Arrowsmith, Holger Braunschweig et Tom E. Stennett, « Formation and Reactivity of Electron-Precise B−B Single and Multiple Bonds », Angewandte Chemie International Edition, vol. 56, no 1, , p. 96–115 (ISSN 1521-3773, PMID 27860056, DOI 10.1002/anie.201610072, lire en ligne)
- Advanced Organic Chemistry, F.A. carey, R.J. Sundberg (ISBN 0-306-41088-5)
- Negishi, E.-i.; Idacavage, M. J, « Formation of Carbon–Carbon and Carbon–Heteroatom Bonds via Organoboranes and Organoborates », Org. React., vol. 33, no 1, , p. 1-246 (DOI 10.1002/0471264180.or033.01)
- Herbert Charles Brown, Michael W. Rathke, « Reaction of carbon monoxide at atmospheric pressure with trialkylboranes in the presence of sodium- or lithium borohydride. A convenient procedure for the oxymethylation of olefins via hydroboration », J. Am. Chem. Soc, vol. 89, no 11, , p. 2740–2741 (DOI 10.1021/ja00987a044)
- Lachance H., Hall D., « Allylboration of Carbonyl Compounds », Org. React., vol. 73, , p. 1 (ISBN 978-0-471-26418-7, DOI 10.1002/0471264180.or073.01)
- Nicolaou, K.C., Sarabia, F., Ninkovic, S., Finlay, M.R.V. et Boddy, C.N.C., « Probing the Ring Size of Epothilones: Total Synthesis of 14-, 15-, 17-, and 18 Epothilones A », Angewandte Chemie International Edition in English, vol. 37, nos 1–2, , p. 81–84 (DOI 10.1002/(sici)1521-3773(19980202)37:1/2<81::aid-anie81>3.0.co;2-c, lire en ligne, consulté le )
- Vedejs E., Chapman R. W., Fields S. C., Lin S., Schrimpf M. R., « Conversion of Arylboronic Acids into Potassium Aryltrifluoroborates: Convenient Precursors of Arylboron Difluoride Lewis Acids », J. Org. Chem., vol. 60, no 10, , p. 3020–3027 (DOI 10.1021/jo00115a016)
- Molander Gary A., Canturk Belgin, « Organotrifluoroborates and Monocoordinated Palladium Complexes as Catalysts—A Perfect Combination for Suzuki–Miyaura Coupling », Angew. Chem. Int. Ed., vol. 48, no 49, , p. 9240–9261 (PMID 19899086, PMCID 2917751, DOI 10.1002/anie.200904306)
- Batey Robert A., Quach Tan D., Shen Ming, Thadani Avinash N., Smil David V., Li Sze-Wan, MacKay D. Bruce, « Organoboron compounds as mild nucleophiles in Lewis acid- and transition metal-catalyzed C–C bond-forming reactions », Pure Appl. Chem., vol. 74, no 1, , p. 43–55 (DOI 10.1351/pac200274010043, lire en ligne)
- Miyaura, Norio et Suzuki, Akira, « Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds », Chemical Reviews, vol. 95, no 7, , p. 2457–2483 (DOI 10.1021/cr00039a007, CiteSeerx 10.1.1.735.7660)
| C-H | He | |||||||||||||||||
| C-Li | C-Be | C-B | C-C | C-N | C-O | C-F | Ne | |||||||||||
| C-Na | C-Mg | C-Al | C-Si | C-P | C-S | C-Cl | C-Ar | |||||||||||
| C-K | C-Ca | C-Sc | C-Ti | C-V | C-Cr | C-Mn | C-Fe | C-Co | C-Ni | C-Cu | C-Zn | C-Ga | C-Ge | C-As | C-Se | C-Br | C-Kr | |
| C-Rb | C-Sr | C-Y | C-Zr | C-Nb | C-Mo | C-Tc | C-Ru | C-Rh | C-Pd | C-Ag | C-Cd | C-In | C-Sn | C-Sb | C-Te | C-I | C-Xe | |
| C-Cs | C-Ba | * | C-Lu | C-Hf | C-Ta | C-W | C-Re | C-Os | C-Ir | C-Pt | C-Au | C-Hg | C-Tl | C-Pb | C-Bi | C-Po | C-At | Rn |
| Fr | C-Ra | * * |
Lr | Rf | Db | C-Sg | Bh | Hs | Mt | Ds | Rg | Cn | Nh | Fl | Mc | Lv | Ts | Og |
| ↓ | ||||||||||||||||||
| * | C-La | C-Ce | C-Pr | C-Nd | C-Pm | C-Sm | C-Eu | C-Gd | C-Tb | C-Dy | C-Ho | C-Er | C-Tm | C-Yb | ||||
| * * |
Ac | C-Th | C-Pa | C-U | C-Np | C-Pu | C-Am | C-Cm | C-Bk | C-Cf | C-Es | Fm | Md | No | ||||
| Liaison de base en chimie organique | Nombreuses utilisations en chimie |
| Recherche académique, mais pas d'usage courant | Liaison inconnue / non évaluée |
 Portail de la chimie
Portail de la chimie