Cystinosis
| Cystinosis | |
|---|---|
| Other names: Cystine storage disease[1] | |
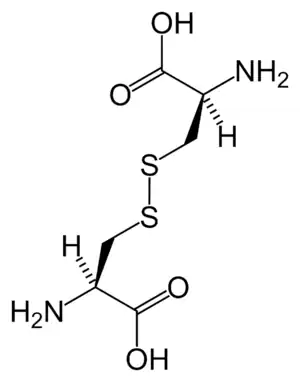 | |
| Chemical structure of cystine formed from L-cysteine (under biological conditions) | |
Cystinosis is a lysosomal storage disease characterized by the abnormal accumulation of cystine, the oxidized dimer of the amino acid cysteine.[2] It is a genetic disorder that follows an autosomal recessive inheritance pattern. It is a rare autosomal recessive disorder resulting from accumulation of free cystine in lysosomes, eventually leading to intracellular crystal formation throughout the body. Cystinosis is the most common cause of Fanconi syndrome in the pediatric age group. Fanconi syndrome occurs when the function of cells in renal tubules is impaired, leading to abnormal amounts of carbohydrates and amino acids in the urine, excessive urination, and low blood levels of potassium and phosphates.
Cystinosis was the first documented genetic disease belonging to the group of lysosomal storage disease disorders.[3] Cystinosis is caused by mutations in the CTNS gene that codes for cystinosin, the lysosomal membrane-specific transporter for cystine. Intracellular metabolism of cystine, as it happens with all amino acids, requires its transport across the cell membrane. After degradation of endocytosed protein to cystine within lysosomes, it is normally transported to the cytosol. But if there is a defect in the carrier protein, cystine is accumulated in lysosomes. As cystine is highly insoluble, when its concentration in tissue lysosomes increases, its solubility is immediately exceeded and crystalline precipitates are formed in almost all organs and tissues.[4]
However, the progression of the disease is not related to the presence of crystals in target tissues. Although tissue damage might depend on cystine accumulation, the mechanisms of tissue damage are not fully understood. Increased intracellular cystine profoundly disturbs cellular oxidative metabolism and glutathione status,[5] leading to altered mitochondrial energy metabolism, autophagy, and apoptosis.[6]
Cystinosis is usually treated with cysteamine, which is prescribed to decrease intralysosomal cystine accumulation.[7] However, the discovery of new pathogenic mechanisms and the development of an animal model of the disease may open possibilities for the development of new treatment modalities to improve long-term prognosis.[3]
Symptoms and signs
There (are) three distinct types of cystinosis each with slightly different symptoms: nephropathic cystinosis, intermediate cystinosis, and non-nephropathic or ocular cystinosis. Infants affected by nephropathic cystinosis initially exhibit poor growth and particular kidney problems (sometimes called renal Fanconi syndrome). The kidney problems lead to the loss of important minerals, salts, fluids, and other nutrients. The loss of nutrients not only impairs growth, but may result in soft, bowed bones (hypophosphatemic rickets), especially in the legs. The nutrient imbalances in the body lead to increased urination, thirst, dehydration, and abnormally acidic blood (acidosis).
By about age two, cystine crystals may also be present in the cornea. The buildup of these crystals in the eye causes an increased sensitivity to light (photophobia). Without treatment, children with cystinosis are likely to experience complete kidney failure by about age ten. With treatment this may be delayed into the patients teens or 20s. Other signs and symptoms that may occur in patients include muscle deterioration, blindness, inability to swallow, impaired sweating, decreased hair and skin pigmentation, diabetes, and thyroid and nervous system problems.
The signs and symptoms of intermediate cystinosis are the same as nephropathic cystinosis, but they occur at a later age. Intermediate cystinosis typically begins to affect individuals around age twelve to fifteen. Malfunctioning kidneys and corneal crystals are the main initial features of this disorder. If intermediate cystinosis is left untreated, complete kidney failure will occur, but usually not until the late teens to mid twenties.
People with non-nephropathic or ocular cystinosis do not usually experience growth impairment or kidney malfunction. The only symptom is photophobia due to cystine crystals in the cornea.
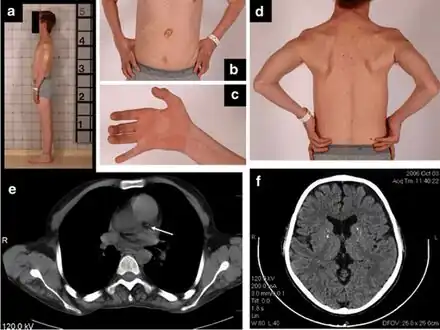 a) Thin habitus b)gastrostomy tube c) clawed hand d) muscle wasting upper trunk e)calcification left coronary artery f) atrophy and calcifications of the basal ganglia
a) Thin habitus b)gastrostomy tube c) clawed hand d) muscle wasting upper trunk e)calcification left coronary artery f) atrophy and calcifications of the basal ganglia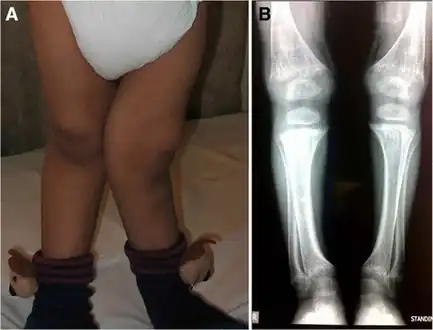 Rickets in cystinosis. a- A cystinosis child with evident rachitic bone deformities. b- Active rachitic bone disease in X-Rays
Rickets in cystinosis. a- A cystinosis child with evident rachitic bone deformities. b- Active rachitic bone disease in X-Rays Slit-lamp photographs of three-year-old patient with nephropathic cystinosis before (left) and after (right) cysteamine eyedrop therapy. The drops dissolve the crystals in the cornea.
Slit-lamp photographs of three-year-old patient with nephropathic cystinosis before (left) and after (right) cysteamine eyedrop therapy. The drops dissolve the crystals in the cornea.
Crystal morphology and identification
Cystine crystals are hexagonal in shape and are colorless. They are not found often in alkaline urine due to their high solubility. The colorless crystals can be difficult to distinguish from uric acid crystals which are also hexagonal. Under polarized examination, the crystals are birefringent with a polarization color interference.[8]
Genetics
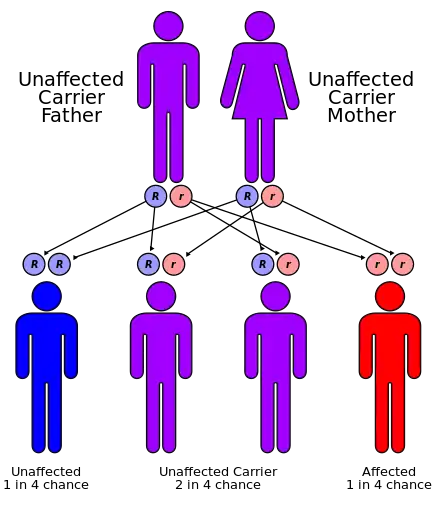
Cystinosis occurs due to a mutation in the gene CTNS, located on chromosome 17, which codes for cystinosin, the lysosomal cystine transporter. Symptoms are first seen at about 3 to 18 months of age with profound polyuria (excessive urination), followed by poor growth, photophobia, and ultimately kidney failure by age 6 years in the nephropathic form.
All forms of cystinosis (nephropathic, juvenile and ocular) are autosomal recessive, which means that the trait is located on an autosomal gene, and an individual who inherits two copies of the gene – one from both parents – will have the disorder. There is a 25% risk of having a child with the disorder, when both parents are carriers of an autosomal recessive trait.
Cystinosis affects approximately 1 in 100,000 to 200,000 newborns.[1] and there are only around 2,000 known individuals with cystinosis in the world. The incidence is higher in the province of Brittany, France, where the disorder affects 1 in 26,000 individuals.[9]
Diagnosis
Cystinosis is a rare genetic disorder[10] that causes an accumulation of the amino acid cystine within cells, forming crystals that can build up and damage the cells. These crystals negatively affect many systems in the body, especially the kidneys and eyes.[2]
The accumulation is caused by abnormal transport of cystine from lysosomes, resulting in a massive intra-lysosomal cystine accumulation in tissues. Via an as yet unknown mechanism, lysosomal cystine appears to amplify and alter apoptosis in such a way that cells die inappropriately, leading to loss of renal epithelial cells. This results in renal Fanconi syndrome,[11] and similar loss in other tissues can account for the short stature, retinopathy, and other features of the disease.
Definitive diagnosis and treatment monitoring are most often performed through measurement of white blood cell cystine level using tandem mass spectrometry.
Types
- Online Mendelian Inheritance in Man (OMIM): 219800 – Infantile nephropathic
- Online Mendelian Inheritance in Man (OMIM): 219900 – Adolescent nephropathic
- Online Mendelian Inheritance in Man (OMIM): 219750 – Adult nonnephropathic
Treatment
Cystinosis is normally treated with cysteamine, which is available in capsules and in eye drops. People with cystinosis are also often given sodium citrate to treat the blood acidosis, as well as potassium and phosphorus supplements as well as others. If the kidneys become significantly impaired or fail, then treatment must be begun to ensure continued survival, up to and including renal transplantation.[12]
Cystinotic
The related adjective "cystinotic" indicates "relating to, or afflicted with, cystinosis".
See also
References
- 1 2 "Cystinosis on Genetic home reference". Archived from the original on 2012-09-10. Retrieved 2021-07-03.
- 1 2 A. Gahl, William; Jess G. Thoene; Jerry A. Schneider (2002). "Cystinosis". N Engl J Med. 347 (2): 111–121. doi:10.1056/NEJMra020552. PMID 12110740.
- 1 2 Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol 2012;28:51–9.
- ↑ Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med 2002;347:111-121.
- ↑ Kumar A, Bachhawat AK. A futile cycle, formed between two ATP-dependent γ-glutamyl cycle enzymes, γ-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis?; J Biosci 2010;35:21–25.
- ↑ Park MA, Thoene JG. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol 2005;20:441–446.
- ↑ Besouw M, Masereeuw R, Van den Heuvel L et al. Cysteamine: an old drug with new potential. Drug Discov Today 2013.
- ↑ Spencer, Daniel. "Cystine". CRYSTALS. Urinalysis (Texas Collaborative for Teaching Excellence). Archived from the original on 6 November 2016. Retrieved 4 March 2012.
- ↑ Kalatzis, V; Cherqui S; Jean G; Cordier B; Cochat P; Broyer M; Antignac C (October 2001). "Characterization of a putative founder mutation that accounts for the high incidence of cystinosis in Brittany". J Am Soc Nephrol. 12 (10): 2170–2174. PMID 11562417. Archived from the original on 14 February 2015. Retrieved 31 March 2011.
- ↑ "Cystinosis". Archived from the original on 2011-07-18. Retrieved 2008-07-20.
- ↑ Howard G. WORTHEN; Robert A. GOOD (1958). "The de Toni-Fanconi Syndrome with Cystinosis". Am J Dis Child. 95 (6): 653–688. doi:10.1001/archpedi.1958.02060050657011. PMID 13532161.
- ↑ Nesterova, Galina; Gahl, William A. (October 6, 2016). "Cystinosis". GeneReviews. University of Washington, Seattle. Archived from the original on April 5, 2011. Retrieved July 3, 2021.
External links
| Classification | |
|---|---|
| External resources |
- Cystinosis at NLM Genetics Home Reference
- GeneReviews/NCBI/NIH/UW entry on Cystinosis Archived 2021-09-25 at the Wayback Machine