D-Glyceric acidemia
| D-Glyceric acidemia | |
|---|---|
| Other names: D-glycerate kinase deficiency[1] | |
 | |
| This condition is inherited in an autosomal recessive manner. | |
D-Glyceric acidemia (D-Glyceric aciduria) is an inherited disease, in the category of inborn errors of metabolism. It is caused by a mutation in the gene GLYCTK, which encodes for the enzyme glycerate kinase.
Signs and symptoms
The presentation of D-glyceric aciduria is consistent with the following:[2]
Pathophysiology
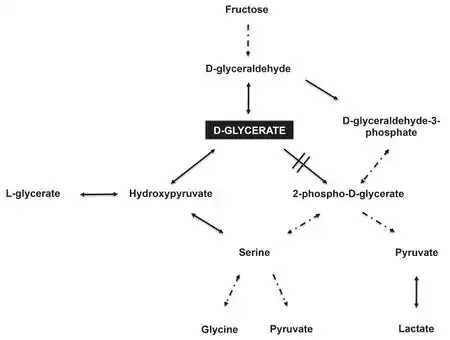
Glycerate kinase is an enzyme that catalyzes the conversion of D-glyceric acid (a.k.a. D-glycerate) to 2-phosphoglycerate. This conversion is an intermediary reaction found in several metabolic pathways, including the degradation (break-down; catabolism) of serine,[3] as well as the breakdown of fructose.[4]
A deficiency in glycerate kinase activity leads to the accumulation of D-glyceric acid (a.k.a. D-glycerate) in bodily fluids and tissues.[5] D-glyceric acid can be measured in a laboratory that performs "analyte testing" for "organic acids" in blood (plasma) and urine.[6]
Symptoms of the disease (in its most severe form) include progressive neurological impairment, mental/motor retardation, hypotonia, seizures, failure to thrive and metabolic acidosis.[7]
Diagnosis
Differential diagnosis
D-Glyceric Acidemia should not be confused with L-Glyceric Acidemia (a.k.a. L-glyceric aciduria, a.k.a. primary hyperoxaluria type II [8]), which is associated with mutations in the GRHPR (encoding for the enzyme 'glyoxylate reductase/hydroxypyruvate reductase').[9][10]
Management
The medical literature indicates there is no current effective treatment, though a fructose-restricted diet has demonstrated a clinical improvement in affected individuals[2]
References
- ↑ "D-Glyceric Acidemia (Concept Id: C1291386) - MedGen - NCBI". www.ncbi.nlm.nih.gov. Retrieved 10 July 2023.
- 1 2 Dimer, Nádia W.; Schuck, Patrícia F.; Streck, Emilio L.; Ferreira, Gustavo C. (August 2015). "D-glyceric aciduria". Anais Da Academia Brasileira De Ciencias. 87 (2 Suppl): 1409–1414. doi:10.1590/0001-3765201520150021. ISSN 1678-2690. Archived from the original on 12 July 2023. Retrieved 10 July 2023.
- ↑ Surtees, Robert; Poll-The, Bwee-Tien; Berger, Ruud; Duran, Marinus; Snell, Keith; Koning, Tom J. de (May 2003). "Biochem. J. (2003) 371, 653-661 - T.J. de Koning and others - l-Serine in disease and development". Biochemical Journal. 371 (3): 653–661. doi:10.1042/bj20021785. PMC 1223326. PMID 12534373.
- ↑ Hommes, F. A. (1993). "Inborn errors of fructose metabolism". Am J Clin Nutr. 58 (5): 788S–795S. doi:10.1093/ajcn/58.5.788S. PMID 8213611. Archived from the original on 2023-07-12. Retrieved 2022-05-28.
- ↑ "GLYCTK - glycerate kinase - Genetics Home Reference". Archived from the original on 2020-09-18. Retrieved 2022-05-28.
- ↑ "GeneTests: Search Results". Archived from the original on 2023-07-12. Retrieved 2022-05-28.
- ↑ Physician's Guide to the Laboratory Diagnosis of Metabolic Diseases. Springer. 2003. ISBN 9783642627095. Archived from the original on 2011-06-06. Retrieved 2022-05-28.
- ↑ "Entry - #260000 - HYPEROXALURIA, PRIMARY, TYPE II; HP2 - OMIM". omim.org. Archived from the original on 9 December 2022. Retrieved 10 July 2023.
- ↑ "Primary Hyperoxaluria Type 2". GeneReviews®. University of Washington, Seattle. 1993. Archived from the original on 2010-06-10. Retrieved 2022-05-28.
- ↑ "OMIM Entry - * 604296 - GLYOXYLATE REDUCTASE/HYDROXYPYRUVATE REDUCTASE; GRHPR". Archived from the original on 2023-07-12. Retrieved 2022-05-28.
External links
| Classification | |
|---|---|
| External resources |
|
- Genetics Home Reference (National Library of Medicine) Archived 2020-09-18 at the Wayback Machine (information on D-glyceric acidemia and the GLYCTK gene)
- OMIM Archived 2023-07-12 at the Wayback Machine (information on GLYCTK gene, encoding Glycerate Kinase)
- GeneTests Archived 2023-07-12 at the Wayback Machine (information on genetic testing for D-Glyceric Acidemia)