Fanconi syndrome
| Fanconi syndrome | |
|---|---|
| Other names: Fanconi's syndrome | |
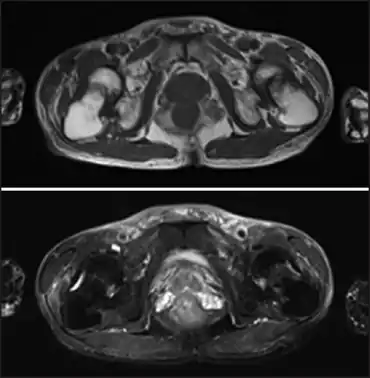 | |
| Fanconi syndrome due to adefovir-image demonstrates low-intensity femoral neck fractures and high-intensity bone edema in both femoral necks | |
Fanconi syndrome or Fanconi's syndrome (English: /fɑːnˈkoʊni/, /fæn-/) is a syndrome of inadequate reabsorption in the proximal renal tubules[1] of the kidney. The syndrome can be caused by various underlying congenital or acquired diseases, by toxicity (for example, from toxic heavy metals), or by adverse drug reactions.[2] It results in various small molecules of metabolism being passed into the urine instead of being reabsorbed from the tubular fluid (for example, glucose, amino acids, uric acid, phosphate, and bicarbonate). Fanconi syndrome affects the proximal tubules, namely, the proximal convoluted tubule (PCT), which is the first part of the tubule to process fluid after it is filtered through the glomerulus, and the proximal straight tubule (pars recta), which leads to the descending limb of loop of Henle.
Different forms of Fanconi syndrome can affect different functions of the proximal tubule, and result in different complications. The loss of bicarbonate results in type 2 or proximal renal tubular acidosis. The loss of phosphate results in the bone diseases rickets and osteomalacia (even with adequate vitamin D and calcium levels), because phosphate is necessary for bone development in children and even for ongoing bone metabolism in adults.[3]
Signs and symptoms
The clinical features of proximal renal tubular acidosis are:
- Polyuria, polydipsia and dehydration
- Hypophosphatemic rickets (in children) and osteomalacia (in adults)
- Growth failure
- Acidosis
- Hypokalemia
- Hyperchloremia
Other features of the generalized proximal tubular dysfunction of the Fanconi syndrome are:
- Hypophosphatemia/hyperphosphaturia
- Glycosuria
- Proteinuria/aminoaciduria
- Hyperuricosuria
Causes
In contrast to Hartnup disease and related tubular conditions, Fanconi syndrome affects the transport of many different substances, so is not considered to be a defect in a specific channel, but a more general defect in the function of the proximal tubules.[4]
Different diseases underlie Fanconi syndrome; they can be inherited, congenital, or acquired.
Inherited
Cystinosis is the most common cause of Fanconi syndrome in children.
Other recognised causes are Wilson's disease (a genetically inherited condition of copper metabolism), Lowe syndrome, tyrosinemia (type I),[5] galactosemia, glycogen storage diseases, and hereditary fructose intolerance.
Two forms, Dent's disease and Lowe syndrome, are X linked.[6]
A recently described form of this disease is due to a mutation in the peroxisomal protein EHHADH.[7] This mutation misdirects the EHHADH to the mitochondria. This interferes with respiratory complex I and with beta oxidation of fatty acids. The end result is a decrease in the ability of the mitochondria to produce ATP.
It was shown that a specific mutation (R76W) of HNF4A, a gene encoding a transcription factor, causes Fanconi syndrome in human.[8] In the kidney, HNF4A is expressed in the proximal tubules specifically.[9] Deletion of Hnf4a in the developing mouse kidney caused Fanconi syndrome phenotypes including polyruia, polydipsia, glycosuria, and phosphaturia.[10] The Hnf4a mutant kidney showed a defect in the formation of proximal tubules.[10]
Acquired
It is possible to acquire this disease later in life.
Causes include ingesting expired tetracyclines (where tetracycline changes to form epitetracycline and anhydrotetracycline which damage the proximal tubule), and as a side effect of tenofovir in cases of pre-existing renal impairment.[11][12] In the HIV population, Fanconi syndrome can develop secondary to the use of an antiretroviral regimen containing tenofovir and didanosine.[13] Lead poisoning also leads to Fanconi syndrome.[14]
Multiple myeloma or monoclonal gammopathy of undetermined significance can also cause the condition.[15]
Additionally, Fanconi Syndrome can develop as a secondary or tertiary effect of certain autoimmune disorders.[16][17]
Diagnosis
Urine routine, might not be completely reliable but is an important indicator.
Treatment
Treatment of children with Fanconi syndrome mainly consists of replacement of substances lost in the urine (mainly fluid and bicarbonate).
Eponym
It is named after Guido Fanconi, a Swiss pediatrician, although various other scientists, including George Lignac, contributed to its study. It should not be confused with Fanconi anemia, a separate disease.
See also
- Familial renal disease in animals for Fanconi syndrome in Basenjis
References
- ↑ "Fanconi syndrome" at Dorland's Medical Dictionary
- ↑ Fanconi Syndrome Archived 2014-11-03 at the Wayback Machine at Merck Manual Home Health Handbook
- ↑ Magen D, Berger L, Coady MJ, Ilivitzki A, Militianu D, Tieder M, Selig S, Lapointe JY, Zelikovic I, Skorecki K (March 2010). "A loss-of-function mutation in NaPi-IIa and renal Fanconi's syndrome". The New England Journal of Medicine. 362 (12): 1102–9. doi:10.1056/NEJMoa0905647. PMID 20335586.
- ↑ Fanconi Syndrome at eMedicine
- ↑ Cochat P, Pichault V, Bacchetta J, Dubourg L, Sabot JF, Saban C, Daudon M, Liutkus A (March 2010). "Nephrolithiasis related to inborn metabolic diseases". Pediatric Nephrology. 25 (3): 415–24. doi:10.1007/s00467-008-1085-6. PMC 2810370. PMID 19156444.
- ↑ Vilasi A, Cutillas PR, Maher AD, Zirah SF, Capasso G, Norden AW, Holmes E, Nicholson JK, Unwin RJ (August 2007). "Combined proteomic and metabonomic studies in three genetic forms of the renal Fanconi syndrome". American Journal of Physiology. Renal Physiology. 293 (2): F456-67. doi:10.1152/ajprenal.00095.2007. PMID 17494094.
- ↑ Assmann N, Dettmer K, Simbuerger JM, Broeker C, Nuernberger N, Renner K, Courtneidge H, Klootwijk ED, Duerkop A, Hall A, Kleta R, Oefner PJ, Reichold M, Reinders J (May 2016). "Renal Fanconi Syndrome Is Caused by a Mistargeting-Based Mitochondriopathy" (PDF). Cell Reports. 15 (7): 1423–1429. doi:10.1016/j.celrep.2016.04.037. PMID 27160910. Archived (PDF) from the original on 2018-07-23. Retrieved 2021-09-19.
- ↑ Hamilton AJ, Bingham C, McDonald TJ, Cook PR, Caswell RC, Weedon MN, Oram RA, Shields BM, Shepherd M, Inward CD, Hamilton-Shield JP, Kohlhase J, Ellard S, Hattersley AT (March 2014). "The HNF4A R76W mutation causes atypical dominant Fanconi syndrome in addition to a β cell phenotype". Journal of Medical Genetics. 51 (3): 165–9. doi:10.1136/jmedgenet-2013-102066. PMC 3932761. PMID 24285859.
- ↑ Lee JW, Chou CL, Knepper MA (November 2015). "Deep Sequencing in Microdissected Renal Tubules Identifies Nephron Segment-Specific Transcriptomes". Journal of the American Society of Nephrology. 26 (11): 2669–77. doi:10.1681/ASN.2014111067. PMC 4625681. PMID 25817355. Archived from the original on 2022-01-21. Retrieved 2021-09-19.
- 1 2 Marable SS, Chung E, Adam M, Potter SS, Park JS (July 2018). "Hnf4a deletion in the mouse kidney phenocopies Fanconi renotubular syndrome". JCI Insight. 3 (14). doi:10.1172/jci.insight.97497. PMC 6124415. PMID 30046000.
- ↑ Viread Label Information Archived 2022-02-20 at the Wayback Machine, U.S. Food and Drug Administration (FDA)), 2008-04-11
- ↑ Tenofovir (Viread) Associated with Mild Kidney Function Impairment, but not Clinically Relevant Renal Disease, hivandhepatitis.com, 2008-10-14
- ↑ Irizarry-Alvarado JM, Dwyer JP, Brumble LM, Alvarez S, Mendez JC (March 2009). "Proximal tubular dysfunction associated with tenofovir and didanosine causing Fanconi syndrome and diabetes insipidus: a report of 3 cases". The AIDS Reader. 19 (3): 114–21. PMID 19334328.
- ↑ Barbier O, Jacquillet G, Tauc M, Cougnon M, Poujeol P (2005). "Effect of heavy metals on, and handling by, the kidney". Nephron Physiology. 99 (4): 105–10. doi:10.1159/000083981. PMID 15722646.
- ↑ Hashimoto T, Arakawa K, Ohta Y, Suehiro T, Uesugi N, Nakayama M, Tsuchihashi T (2007). "Acquired fanconi syndrome with osteomalacia secondary to monoclonal gammopathy of undetermined significance". Internal Medicine. 46 (5): 241–5. doi:10.2169/internalmedicine.46.1882. PMID 17329920.
- ↑ "Fanconi Syndrome". The Medical Dictionary. Archived from the original on 2017-10-10. Retrieved 2021-09-19.
- ↑ Kobayashi T, Muto S, Nemoto J, Miyata Y, Ishiharajima S, Hironaka M, Asano Y, Kusano E (June 2006). "Fanconi's syndrome and distal (type 1) renal tubular acidosis in a patient with primary Sjögren's syndrome with monoclonal gammopathy of undetermined significance". Clinical Nephrology. 65 (6): 427–32. doi:10.5414/CNP65427. PMID 16792139.
External links
| Classification | |
|---|---|
| External resources |