POEMS syndrome
| POEMS syndrome | |
|---|---|
| Other names: Crow–Fukase syndrome | |
POEMS syndrome (also termed osteosclerotic myeloma, Crow–Fukase syndrome, Takatsuki disease, or PEP syndrome) is a rare paraneoplastic syndrome caused by a clone of aberrant plasma cells. The name POEMS is an acronym for some of the disease's major signs and symptoms (polyneuropathy, organomegaly, endocrinopathy, myeloma protein, and skin changes), as is PEP (polyneuropathy, endocrinopathy, plasma cell dyscrasia).
The signs and symptoms of most neoplasms are due to their mass effects caused by the invasion and destruction of tissues by the neoplasms' cells. Signs and symptoms of a cancer causing a paraneoplastic syndrome result from the release of humoral factors such as hormones, cytokines, or immunoglobulins by the syndrome's neoplastic cells and/or the response of the immune system to the neoplasm. Many of the signs and symptoms in POEMS syndrome are due at least in part to the release of an aberrant immunoglobulin, i.e. a myeloma protein, as well as certain cytokines by the malignant plasma cells.[1][2][3]
POEMS syndrome typically begins in middle age – the average age at onset is 50 – and affects up to twice as many men as women.
Signs and symptoms
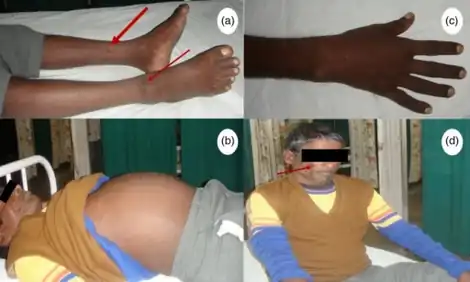
The signs and symptoms of POEMS syndrome are highly variable. This often leads to long delays (e.g. 13–18 months) between the onset of initial symptoms and diagnosis.[1][2] In addition to the signs and symptoms indicated by the POEMS acronym, the PEST acronym is used to describe some of the other signs and symptoms of the disease. PEST stands for Papilledema, evidence of Extravascular volume overload (ascites, pleural effusion, pericardial effusion, and lower extremity edema), Sclerotic bone lesions, and Thrombocytosis/erythrocytosis (i.e. increased in blood platelets and red blood cells). Other features of the disease include a tendency toward leukocytosis, blood clot formation, abnormal lung function (restrictive lung disease, pulmonary hypertension, and impaired lung diffusion capacity), very high blood levels of the cytokine vascular endothelial growth factor (VEGF), and an overlap with the signs and symptoms of multicentric Castleman disease.[2]
Common features
The more common features of the disease are summarized in the acronym POEMS: Papilledema (swelling of the optic disc) often but not always due to increased intracranial pressure) is the most common ocular sign of POEMS syndrome, occurring in ≥29% of cases. Less frequent ocular findings include cystoid macular edema, serous macular detachment, infiltrative orbitopathy, and venous sinus thrombosis.[4]
Pulmonary disease/ Polyneuropathy: The lungs are often affected at more severe stages of the illness, although since by then physical exertion is usually limited by neuropathy, shortness of breath is unusual. Pulmonary hypertension is the most serious effect on the lungs, and there may also be restriction of chest expansion or impaired gas exchange.[2][3]
Organomegaly: The liver may be enlarged, and less often the spleen or lymph nodes, though these organs usually function normally.[2][3]
Edema: Leakage of fluid into the tissues is a common and often severe problem. This may take several forms, including dependent peripheral edema, pulmonary edema, effusions such as pleural effusion or ascites, or generalized capillary leakage (anasarca).[2][3]
Endocrinopathy: In women, amenorrhea, and in men, gynecomastia, erectile dysfunction and testicular atrophy, are common early symptoms due to dysfunction of the gonadal axis. Other hormonal problems occurring in at least a quarter of patients include type 2 diabetes, hypothyroidism, and adrenal insufficiency.[2][3]
Monoclonal paraprotein: In most cases a serum myeloma protein can be detected, although this is not universal. This may represent IgG or IgA, but the light chain type is almost always lambda. This is in contrast to most paraproteinemic neuropathies, in which the paraprotein is usually an IgM antibody.[1]
Skin changes: A very wide variety of skin problems have been reported in association with POEMS syndrome. The most common is non-specific hyperpigmentation. The fingernails may be clubbed or white. There may be thickening of the skin, excess hair or hair in unusual places (hypertrichosis), skin angiomas or hemangiomas, or changes reminiscent of scleroderma.[5]
Possible features
Some features have been observed in patients with POEMS syndrome but are not yet certain to form part of the syndrome itself. These include a predisposition to forming blood clots, joint pain, cardiomyopathy (systolic dysfunction), fever, low vitamin B12 levels, and diarrhea.[6]
Pathogenesis
While the main features of this paraneoplastic disease have been described, the exact mechanism behind its development, progression, and manifestations remain elusive. Overproduction of the myeloma protein and VEGF may underlie some, but are insufficient to explain all, of the multi-organ features of the disease. It is suggested that various other cytokines produced by the clonal plasma cells, perhaps working in concert with each other as well as with VEGF and the myeloma proteins, mediate many of the features of POEMS syndrome. The other cytokines detected in, and suspected of contributing to, POEMS syndrome include interleukin 1β, interleukin 6, and TNFα. Nonetheless, it seems likely that some of these paraneoplastic factors, operating individually, make a major contribution to certain features of the disease. For example, VEGF, given its ability to stimulate blood vessel formation, would seem likely to be the major contributor to the pathologic hyper-vascularization changes seem in many tissues, such as lymph nodes, afflicted by POEMS syndrome.[6]
Diagnosis
The diagnosis of POEMS syndrome is based on meeting its two mandatory criteria, meeting at least one of its 3 other major criteria, and meeting at least one of its 6 minor criteria. These criteria are:[7]
- Mandatory major criteria
- Plasma cell dyscrasia: This is evidenced by 1) the presence of a serum myeloma protein, typically an IgG or IgA isotype (occurs in nearly 100% of cases; in >95% of instances the myeloma proteins contain a λ chain that is restricted to either of two V lambda 1 subfamily members viz., IGLV1-40*01 and IGLV1-44*01 (see V lambda family); 2) any, but often a small, increase above the normal value of <1.5% in the percentage of nucleated bone marrow cells that are clonal plasma cells (occurs ~67% of cases); and/or 3) presence of a plasma cell tumor (i.e. plasmacytoma) usually in bone (occurs in ~33% of cases).[2]
- Polyneuropathy: The nerve damage is usually symmetrical, located in distal extremities, and due to the nerve losing its fatty myelin coating and axonal damage. Neurons of the Sensory, motor and autonomic nervous systems are all affected. The typical symptoms are therefore numbness, tingling, and weakness in the feet, later affecting the legs and hands. Pain is unusual, but the weakness may eventually become severe and disabling. The autonomic neuropathy may cause excessive sweating and erectile dysfunction; hormonal changes may also contribute to the latter. It is usually the symptoms of neuropathy which prompt a person with POEMS syndrome to seek medical attention.[8]
- Other major criteria
- Castleman disease: The lymphoproliferative disorder Castleman disease associated with POEMS syndrome is multicentric and occurs in ~15 of cases. It is characterized by a morphology in lymph nodes termed angiofollicular lymph node hyperplasia; an overly activate immune system; excessive production of cytokines including particularly IL-6 and to lesser extents, proliferation of immune B cells and T cells, enlarged lymph nodes, enlarged liver and spleen, capillary leak syndrome, anasarca, evidence of extravascular fluid overload, and organ failure. Patients with Castleman disease without a plasma cell dyscrasia and peripheral neuropathy but having other signs and symptoms of POEMS syndrome can be classified as a Castleman disease variant of POEMS syndrome.[2]
- Sclerotic bone lesions: These lesions consist of plasma cell tumors encased within or associated with abnormally dense bone structures; in different studies, they have been observed to occur in 27% to 97% of cases.[2][7]
- Elevated VEGF: VEGF is a cytokine that stimulates angiogenesis (i.e. capillary formation), increases capillary permeability, and contributes to polyneuropathy. It is elevated in almost all cases of POEMS syndrome and has become a clinically useful marker for the syndrome's presence, severity, and response to treatment. However, its role in mediating the symptoms of this disease are unclear. A second cytokine, IL-12, is similar to VEGF in being highly correlated with the disease activity level in POEMS syndrome.[2]
- Minor criteria
- Organomegaly: Enlargement spleen, liver, and/or lymph nodes occurs in 45% to 85% of cases.[7]
- Extravascular volume overload: Ascites, pleural effusions, pericardial effusions, and/or lower extremity edema occur in 27% to 89% of cases.[7]
- Endocrinopathy: Gynecomastia occurs in 12% to 18% of cases; endocrine abnormalities involving the regulation of gonadotrophins, adrenal gland corticosteroids, and prolactin occur in 55% to 89%, 16% to 33%, and 55 to 20% of cases, respectively. Diabetes and hypothyroidism also occur in 3% to 36% and 9% to 67%, respectively, of cases but are not considered to be criteria for the presence of POEMS syndrome because of their frequent occurrence in the general population.[7]
- Skin changes: Skin changes occur in 68% to 89% of POEMS syndrome patients. These changes most commonly are hyperpigmentation and/or hypertrichosis (abnormal amount of hair growth over the body) but less commonly include glomeruloid hemangioma, signs or symptoms of Hypervolemia (e.g. edema and ascites), acrocyanosis (blue discoloration of the extremities due to blood flow abnormalities), flushing, and/or white nails.[3]
- Papilledema: Papilledema (swelling of retinal optical discs) occurs in 29% to 64% of cases. Papilledema in POEMS syndrome patients may occur with or without visual disturbances, increased intracranial pressure, or changes in cerebral spinal fluid protein levels.[3][9]
- Thrombocytosis/polycythemia: Thrombocytosis (increase in blood platelet count) and polycythemia (increase in red blood cells) occurs 54% to 88% and 12% to 19%, respectively, of POEMS syndrome patients and may be may underlying causes of these patients to experience thrombosis events.[3]
Laboratory findings
In addition to tests corresponding to the above findings (such as EMG for neuropathy, CT scan, bone marrow biopsy to detect clonal plasma cells, plasma or serum protein electrophoresis to myeloma proteins, other tests can give abnormal results supporting the diagnosis of POEMS syndrome. These included raised blood levels of VEGF, thrombocytes, and/or erythrocyte parameters.[2]
Differential diagnosis
Patients diagnosed as having Castleman disease but also exhibiting many of the symptoms and signs of POEMS syndrome but lacking evidence of a peripheral neuropathy and/or clonal plasma cells should not be diagnosed as having POEMS syndrome. They are better classified as having Castleman disease variant of POEMS syndrome. These patients may exhibit high blood levels of the interleukin-6 cytokine and have an inferior overall survival compared to POEMS syndrome patients. Treatment of patients with this POEMS syndrome variant who have evidence of bone lesions and/or myeloma proteins are the same as those for POEMS syndrome patients. In the absence of these features, treatment with rituximab, a monoclonal antibody preparation directed against B cells bearing the CD20 antigen, or siltuximab, a monoclonal antibody preparation directed against interleukin-6, may be justified.[2][3]
Treatment
As reported by Dispenzieri et al.[3] Mayo Clinic treatment regimens are tailored to treat the clinical manifestations and prognosis for the rate of progression of the POEMS syndrome in each patient. In rare cases, patients may have minimal or no symptoms at presentation or after successful treatment of their disorder. These patients may be monitored every 2–3 months for symptoms and disease progression. Otherwise, treatment is divided based on the local versus systemic spread of its clonal plasma cells. Patients with one or two plasmacytoma bone lesions and no clonal plasma cells in their bone marrow biopsy specimens are treated by surgical removal or radiotherapy of their tumors. These treatments can relieve many of the syndromes clinical manifestations including neuropathies, have a 10-year overall survival of 70% and a 6-year progression-free survival of 62%. Patients with >2 plasmacytoma bone lesions and/or increases in bone marrow clonal plasma cells are treated with a low-dose or high-dose chemotherapy regimen, i.e. a corticosteroid such as dexamethasone plus an alkylating agents such as melphalan. Dosage regimens are selected on the basis of patient tolerance. Hematological response rates to the dexamethasone/melphalan regimens have been reported to be in the 80% range with neurological response rates approaching 100%. Patients successfully treated with the high-dose dexamethasone/melphalan regimen have been further treated with autologous stem cell transplantation. In 59 patients treated with the chemotherapy/transplantation regimen, the Mayo Clinic reported progression-free survival rates of 98%, 94%, and 75% at 1, 2, and 5 years, respectively.[3]
Other treatment regiments are being studied. Immunomodulatory imide drugs such as thalidomide and lenalidomide have been used in combination with dexamethasone to treat POEMS syndrome patients. While the mechanism of action of these immunomodulators are not clear, they do inhibit the production of cytokines suspected of contributing to POEMS syndrome such as VEGF, TNFα, and IL-6 and stimulate T cells and NK cells to increase their production of interferon gamma and interleukin 2 (see immunomodulatory imide drug's mechanism of action). A double blind study of 25 POEMS syndrome patients found significantly better results (VEGF reduction, neuromuscular function improvement, quality of life improvement) in patients treated with thalidomide plus dexamethasone compared to patients treated with a thalidomide placebo plus dexamethasone.[2]
Since VEGF plays a central role in the symptoms of POEMS syndrome, some have tried bevacizumab, a monoclonal antibody directed against VEGF. While some reports were positive, others have reported capillary leak syndrome suspected to be the result of overly rapid lowering of VEGF levels. It therefore remains doubtful as to whether this will become part of standard treatment for POEMS syndrome.[10]
History
R. S. Crow, working in Bristol, first described the combination of osteosclerotic myeloma, polyneuropathy and various unusual features (such as pigmentation and clubbing) in two patients aged 54 and 67.[11]
References
- 1 2 3 Castillo JJ (2016). "Plasma Cell Disorders". Primary Care. 43 (4): 677–691. doi:10.1016/j.pop.2016.07.002. PMID 27866585.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Warsame R, Yanamandra U, Kapoor P (2017). "POEMS Syndrome: an Enigma". Current Hematologic Malignancy Reports. 12 (2): 85–95. doi:10.1007/s11899-017-0367-0. PMID 28299525. S2CID 31324035.
- 1 2 3 4 5 6 7 8 9 10 11 Dispenzieri A (2017). "POEMS syndrome: 2017 Update on diagnosis, risk stratification, and management". American Journal of Hematology. 92 (8): 814–829. doi:10.1002/ajh.24802. PMID 28699668.
- ↑ Kaushik M, Pulido JS, Abreu R, Amselem L, Dispenzieri A (2011). "Ocular findings in patients with polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes syndrome". Ophthalmology. 118 (4): 778–82. doi:10.1016/j.ophtha.2010.08.013. PMID 21035860.
- ↑ Dispenzieri A, Buadi FK (December 2013). "A review of POEMS syndrome". Oncology (Williston Park, N.Y.). 27 (12): 1242–50. PMID 24624542. Archived from the original on 2019-01-22. Retrieved 2021-08-11.
- 1 2 Dispenzieri A, Kyle RA, Lacy MQ, et al. (2003). "POEMS syndrome: definitions and long-term outcome". Blood. 101 (7): 2496–506. doi:10.1182/blood-2002-07-2299. PMID 12456500. Archived from the original on 2009-01-25. Retrieved 2008-04-13.
- 1 2 3 4 5 Dispenzieri A (2014). "POEMS syndrome: 2014 update on diagnosis, risk-stratification, and management". American Journal of Hematology. 89 (2): 214–23. doi:10.1002/ajh.23644. PMID 24532337. S2CID 33370434.
- ↑ Rosenbaum E, Marks D, Raza S (2017). "Diagnosis and management of neuropathies associated with plasma cell dyscrasias". Hematological Oncology. 36 (1): 3–14. doi:10.1002/hon.2417. PMID 28397326. S2CID 3440992.
- ↑ Lam C, Margolin E (2016). "A case of POEMS and chronic papilledema with preserved optic nerve function". Canadian Journal of Ophthalmology. 51 (1): e8–10. doi:10.1016/j.jcjo.2015.08.015. PMID 26874176.
- ↑ Samaras P, Bauer S, Stenner-Liewen F, et al. (2007). "Treatment of POEMS syndrome with bevacizumab". Haematologica. 92 (10): 1438–9. doi:10.3324/haematol.11315. PMID 18024383. Archived from the original on 2011-10-02. Retrieved 2021-08-11.
- ↑ Crow RS (1956). "Peripheral neuritis in myelomatosis". Br Med J. 2 (4996): 802–4. doi:10.1136/bmj.2.4996.802. PMC 2035359. PMID 13364332.
External links
| Classification | |
|---|---|
| External resources |