Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is a cancer of B cells, a type of lymphocyte that is responsible for producing antibodies. It is the most common form of non-Hodgkin lymphoma among adults,[1] with an annual incidence of 7–8 cases per 100,000 people per year in the US and UK.[2][3] This cancer occurs primarily in older individuals, with a median age of diagnosis at ~70 years,[3] although it can occur in young adults and, in rare cases, children.[4] DLBCL can arise in virtually any part of the body and, depending on various factors, is often a very aggressive malignancy.[5] The first sign of this illness is typically the observation of a rapidly growing mass or tissue infiltration that is sometimes associated with systemic B symptoms, e.g. fever, weight loss, and night sweats.[6]
| Diffuse large B cell lymphoma | |
|---|---|
| Other names | DLBCL or DLBL |
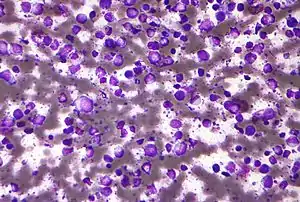 | |
| Micrograph (Field stain) of a diffuse large B cell lymphoma. | |
| Specialty | Hematology, oncology, dermatology |
The causes of diffuse large B-cell lymphoma are not well understood. Usually DLBCL arises from normal B cells, but it can also represent a malignant transformation of other types of lymphoma (particularly marginal zone lymphomas[7]) or, in rare cases termed Richter's transformation, chronic lymphocytic leukemia.[8] An underlying immunodeficiency is a significant risk factor for development of the disease.[9] Infections with the Epstein–Barr virus (EBV),[10][11] Kaposi's sarcoma-associated herpesvirus,[12][13] human immunodeficiency virus (i.e. HIV),[12] and the Helicobacter pylori bacterium [7] are also associated with the development of certain subtypes of diffuse large B-cell lymphoma. However, most cases of this disease are associated with the unexplained step-wise acquisition of increasing numbers of gene mutations and changes in gene expression that occur in, and progressively promote the malignant behavior of, certain B-cell types.[14]
Diagnosis of DLBCL is made by removing a portion of the tumor through a biopsy, and then examining this tissue using a microscope. Usually a hematopathologist makes this diagnosis.[15] Numerous subtypes of DLBCL have been identified which differ in their clinical presentations, biopsy findings, aggressive characteristics, prognoses, and recommended treatments.[16] However, the usual treatment for most subtypes of DLBCL is chemotherapy combined with a monoclonal antibody drug that targets the disease's cancerous B-cells, usually rituximab.[17] Through these treatments, more than half of all patients with DLBCL can be cured;[18] the overall cure rate for older adults is less than this but their five-year survival rate has been around 58%.[19]
Subtypes of diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma encompasses a biologically and clinically diverse set of disease subtypes,[20] many of which are difficult to separate from one another based on well-defined and widely accepted criteria. The World Health Organization, 2008, classification system defined more than a dozen subtypes,[21] each of which was identified based on the location of the tumor, the presence of other cell types such as T cells in the tumor, and whether the patient had certain other illnesses related to DLBCL. Based on further research, the World Health Organization, 2016, reclassified DLBCL into its most common subtype, diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS). DLBCL, NOS represents 80–85% of all DLBCL.[22] The remaining DLBCL cases consist of relatively rare subtypes that are distinguished by their morphology, (i.e. microscopic appearance), immunophenotype, (i.e. expression of certain marker proteins), clinical findings, and/or association with certain pathogenic viruses.[12] Some cases of DLBCL, NOS, while not included in the 2016 World Health Organization's classification, are clearly associated with, and caused by, chronic infection by the bacterium, Helicobacter pylori.[23]
Diffuse large B-cell lymphoma, not otherwise specified
DLBCL cases that do not fit the distinctive clinical presentation, tissue morphology, neoplastic cell phenotype, and/or pathogen-associated criteria of other DLBCL subtypes are termed Diffuse large B-cell lymphoma, not otherwise specified: DLBCL, NOS, while representing 80–85% of all DLBCL cases, is a diagnosis of exclusion. In general, DLBCL, NOS is an aggressive disease with an overall long-term survival rate in patients treated with standard chemotherapy regimens of ~65%. However, this disease has many variants that differ not only in the just cited parameters but also in their aggressiveness and responsiveness to treatment.[24]
Presenting signs and symptoms
About 70% of DLBCL, NOS cases present primarily with lymph node disease. In these cases, the most typical presenting symptom at the time of diagnosis is a mass that is rapidly enlarging and located in a part of the body with multiple lymph nodes such as the groin, arm pits, or neck. In the remaining ~30% of other cases, the disease begins as an extranodal lymphoma, most commonly in the stomach,[12] or, less commonly, in other sites such as the testicles, breasts, uterus, ovaries, kidneys, adrenal glands, thyroid gland, or bone.[25] The presenting signs and symptoms in these cases reflect the presence of a rapidly expanding tumor or infiltrate that produces symptoms specific to the organ of involvement such as increased size, pain, and/or dysfunction.[25] Individuals with nodal or extranodal disease also present with: systemic B symptoms such as weight loss, night sweats, fevers, and/or fatigue in ~33% of cases; unexplained elevations in their blood levels of lactic acid dehydrogenase and beta-2 microglobulin in many cases; malignant cells infiltrating their bone marrow in 10–20% of cases; and/or localized Stage I or II disease in up to 50% of cases and disseminated Stage III or IV disease in the remaining cases.[12] Bone marrow involvement may be due to DLBCL, NOS cells or low grade lymphoma cells; only DLBCL, NOS cell infiltrates indicate a worse prognosis.[22] Uncommonly, DLBCL may arise as a transformation of marginal zone lymphoma (MZL) in individuals who have been diagnosed with this indolent cancer 4–5 years (median times) previously.[26]
Prognostic indicators based on clinical presentation
The International Prognostic Index and more recently, the Index's age-adjusted variant use age >60 years, elevated serum lactate dehydrogenase levels, low performance status, and involvement in more than one extranodal site as contributors to a poor prognosis in patients with DLBCL, NOS.[22] In addition, disease that initially involves the testes, breast, or uterus has a relatively high rate of spreading to the central nervous system while disease initially involving the kidneys, adrenal glands, ovaries, or bone marrow has a high rate of spreading to other organs, including the central nervous system. All of these cases as well as cases initially involving the central nervous system have relatively poor to very poor prognoses. Cases initially involving the stomach, thyroid, or a single bone site have relatively good prognoses.[25]
Pathophysiology
Most cases of DLBCL, NOS appear to result at least in part from the step-wise development of gene changes such as mutations, altered expressions, amplifications (i.e. increases in the number of copies of specific genes), and tranlocations from normal sites to other chromosomal sites. These changes often result in gains or loses in the production or function of the product of these genes and thereby the activity of cell signaling pathways that regulate the maturation, proliferation, survival, spread, evasion of the immune system, and other malignant behaviors of the cells in which they occur. While scores of genes have been reported to be altered in DLBCL, NOS many of these may not contribute to DLBCL, NOS. Changes in the following genes occur frequently in, and are suspected of contributing to, this disease's development and/or progression.[14]
- BCL2: This gene is a protooncogene, i.e. a normal gene that can become cancer-causing when mutated or overexpressed. Its product, Bcl-2 protein, regulates cellular apoptosis (i.e. survival) by inhibiting the apoptosis-causing proteins, Bcl-2-associated X protein and Bcl-2 homologous antagonist killer.[27]
- BCL6: This genes' product, Bcl-6, is a repressor of transcription that regulates the expression of other genes which control cell maturation, proliferation, and survival.[27]
- MYC: This protooncogene's product, Myc, encodes a transcription factor which regulates the expression of other genes whose products stimulate cell proliferation and expansion to extra-nodal tissues.[28]
- EZH2: This gene's product, the EZH2 protein, is a histone-lysine N-methyltransferase. It thereby regulates the expression of other genes which control lymphocyte maturation.[22]
- MYD88: This gene's product is a signal transducing adaptor protein essential for the transduction of interleukin-1 and toll-like receptor signaling pathways. It thereby regulates NF-κB and MAPK/ERK signaling pathways that control cell proliferation and survival.[27]
- CREBBP: This gene's product is a transcriptional coactivator; it activates numerous transcription factors, some of which control cell proliferation.[14]
- CD79A and CD79B: these genes' products are critical components of the B-cell receptor. Mutations in either gene can cause uncontrolled cell activation and proliferation.[14]
- PAX5: this gene's product, Pax-5, is a transcription factor that controls the development, maturation, and survival of B-cells; it also controls expression of the MYC gene in these cells.[29]
As a consequence of these gene changes and possibly other changes that have not yet been identified, the neoplastic cells in DLBCL, NOS exhibit pathologically overactive NF-κB, PI3K/AKT/mTOR, JAK-STAT0, MAPK/ERK, B-cell receptor, toll-like receptor, and NF-κB signaling pathways and thereby uncontrolled pro-malignant behaviors.[27]
Diagnosis
Microscopic examinations of involved tissues reveal large neoplastic cells that are typically classified as B-cells based on their expression of B-cell marker proteins (e.g. CD20, CD19, CD22, CD79, PAX5, BOB1, OCT2, an immunoglobulin [usually IgM but occasionally IgG or IgA)],[12] CD30, and in ~20–25% of cases PD-L1 or PD-L2 (PD-L1 and PD-L2 are transmembrane proteins that normally function to suppress attack by the immune system).[22] These cells arrange in a diffuse pattern, efface the tissues' architecture, and resemble Centroblast cells (80% of cases), Immunoblast cells (8–10% of cases), or anaplastic cells (9% of cases; anaplastic cells have bizarre nuclei and other features that may mimic the Reed–Sternberg cells of Hodgkin disease or the neoplastic cells of anaplastic large cell lymphoma). Rarely, these neoplastic cells are characterized as having signet ring or spindle shaped nuclei, prominent cytoplasmic granules, multiple microvillus projections, or, when viewed by electron microscopy, tight junctions with other cells.[12] These neoplastic tissue infiltrates are often accompanied by small non-malignant T-cell lymphocytes and histiocytes that have a reactive morphology.[22]
Variants of DLBCL, NOS
The World Health Organization, 2016, requires that the neoplastic cells in DLBCL, NOS be further defined based on whether they are derived from germinal center B-cells (i.e. GBC) or activated B-cells (i.e. ABC) as identified by gene expression profiling (GEP) or are GBC or non-GBC as identified by immunohistochemical (IHC) analyses. As identified by GEP, which measures all cellular messenger RNAs, GBC and ABC represent about 50 and ~35% of DLBCL, NOS cases, respectively, with ~15% of cases being unclassifiable.[30] IHC analyses measure the cellular expression of specific proteins using a panel of fluorescent antibodies that bind to and therefore stain a set of key proteins. For example, one commercially available panel uses three antibodies to detect CD10, BCL6, and MUM1 proteins; GBC express whereas ABC and unidentified cells do not express these proteins; accordingly, this as well as other IHC panels classify ABC and undetermined neoplastic cell types together as non-GBC.[27] Individuals with the ABC, unclassifiable, and non-GBC variants have significantly worse prognoses than individuals with the GBC variant:[24] respective 5 year progression-free and overall survival rates have been reported to be 73–80% for GBC variants and 31–56% for ABC variants. Clinically, however, most DLBCL, NOS cases are analyzed by IHC and therefore classified as either GBC or non-GBC variants with non-GBC variants having progression-free and overall survival rates similar to those of the ABC variants.[22]
Gene and protein markers in the neoplastic cells of DLBCL, NOS that have clinical significance include CD5, MYC, BCL2, BCL6,[12] CD20, CD19, CD22, CD30, PD-L1, and PD-L2.[24] The 5–10% of DLBCL, NOS cases in which the neoplastic cells express CD5 have a very poor prognosis that is not improved by even aggressive treatment regimens. Cases in which fluorescence in situ hybridization analysis show that the neoplastic cells' in this disease bear translocations in both the MYC and BCL2 genes or MYC and BCL6 genes (termed double hit lymphomas) or in all three genes (termed triple hit lymphomas)[22] are associated with advanced disease that spreads to the central nervous system.[28] These lymphomas, termed high-grade B-cell lymphoma with MYC, BL2, and/or BL6 rearrangements or, more simply, DH/THL, are regarded as borderline DLBCL,NOS.[22] They represent 6–14% of all DLBCL, NOS and have had long-term survival rates of only 20–25%.[25] Another variant B-cell lymphoma that is also considered to be a borderline DLBCL, NOS is termed high-grade B-cell lymphoma, not otherwise specified (HGBCL, NOS).[22] These two aggressive borderline B-cell lymphomas were previously grouped together as "B-cell lymphoma, unclassifiable with features intermediate between DLBCL and Burkitt lymphoma" (i.e. BCLU) but were separated into DH/THL and HGBC, NOS by the World Health Organization, 2016.[16] The neoplastic cells in a related variant, double expresser lymphoma (i.e. DEL), express the products of MYC and BCL2 genes, i.e. c-Myc and bcl-2 proteins, respectively, but do not have translocations in either of their genes. DEL, which represents about one-third of all DLBCL, NOS cases, has a poorer prognosis than standard DLBCL, NOS but not as poor as DH/THL cases.[22][30] Cases in which the neoplastic cells have alterations in the MYC gene or its expression without changes in BLC2 or BLC6 also have a poor prognosis,[22] particularly in cases where the MYC gene translocates (i.e. rearranges) with one of the immunoglobulin gene loci. DLBCL that begin in the testicles are a variant of DLBCL, NOS that some authors suggest should be classified as a distinct DLBCL subtype.[12] This variant, termed Primary testicular diffuse large B-cell lymphoma (PT-DLBCL), is a DLBCL, NOS that in >75% of cases involves activated B-cells, i.e. ABC.[31] These cells, which typically have a centroblast-like morphology, infiltrate one or, in ~6% of cases, both testicles. PT-DLBCL is an aggressive disease that often spreads to the central nervous system[31] and has median overall survival and progression-free survival times of 96 and 49 months, respectively.[12]
The neoplastic cells in almost all cases of DLBCL, NOS express CD20. Commercially available anti-CD20 antibody agents such as rituximab or Obinutuzumab (which is sometimes used in place of rituximab) kill cells that express high levels of CD20 by binding to this cell-surface protein and thereby targeting them for attack by the hosts adaptive immune system. The addition of one of these immunotherapy agents to chemotherapy protocols has greatly improved the prognosis of most DLBCL, NOS variants.[22] Neoplastic cell expression of CD30, found in 10–15% of DLBCL, NOS cases is a favorable prognostic indicator. As indicated in the following Treatments and prognoses section, expression of the CD20 and CD30 proteins as well as the CD19, CD20 CD22, CD30, CD79A, CD79B, and D-L1 proteins, expression of the MYC, BCL2, MYD88nd, and CREBBP genes, and expression of the PI3K/AKT/mTOR, JAK-STAT, B-cell receptor, toll-like receptor, and NF-κB signaling pathways are being studied as potential therapeutic targets for the individualized treatment of GBC and ABC/non-GBC DLBCL, NOS cases.[14][22]
First-line therapy
First-line therapy for patients with the GBC variant of DLBCL, NOS is R-CHOP. R-CHOP consists of rituximab, three chemotherapy drugs (cyclophosphamide, doxorubicin, and vincristine) and a glucocorticoid (either prednisone or prednisolone).[30] The regimen achieves cure, relapse following remission,[22] and unresponsive rates of 60–70%, 30–40% and <10%, respectively, in GBC variant cases.[32] Relapses generally occur within the first 3 years of diagnosis with few cases doing so after 5 years. Patients who are refractory to, relapse within 1 year of diagnosis before starting, relapse within 6 months after completing, or progress within 2 years of starting R-CHOP have poorer prognoses.[28] R-CHOP is less effective and not recommended for patients who have MYC, BL2, and/or BL6 rearrangements regardless of their GBC, ABC, or non-GBC type. One recommendation for treating these DH/THL cases is the DA-R-EPOCH regimen (dose-adjusted rituximab, etoposide, prednisolone, oncovin, cyclophosphamide, and hydroxydaunorubicin). S-R-EPOCH achieves 2 year survival rates of 40–67% compared to a ~25% survival rate for R-CHOP in these cases.[30] DA-R-EPOCH has also been recommended for patients with double expresser lymphoma[30] although some experts recommend treating this variant more like a typical DLCBL, NOS.[27] First-line therapy for patients with the ABC, undetermined, or non-GBC variants has been the DA-R-EPOCH regimen. Patients with these variants (including those with double expresser lymphoma) have had a ~40% cure rate when treated with it.[30] A randomized clinical trial conducted in France reported that a R-ACVBP chemotherapy regimen (rituximab, adriamycin, cyclophosphamide, vindesine, bleomycin, and cytarabine followed by sequential consolidation therapy with systemic methotrexate, ifosfamide, and etoposide, and then cytarabine) achieved significantly better response rates than R-CHOP in ABC/NGC variant cases lymphoma.[27] In DLBCL, NOS variants which trend to spread or to the central nervous system, methotrexate has been recommended to be added to regimens not containing it for use as prophylaxis to reduce the incidence of this complication.[28] The role of Autologous stem-cell transplantation as an addition to first-line therapy in the treatment of DLBCL, NOS, including cases with a poor prognosis, is unclear.[14]
A phase I clinical research trial found that the addition of lenalidomide to the R-CHOP regimen produce an ~80% complete response rate in GBC as well as non- GBC DLBCL, NOS variants.[14] Two phase III clinical research trials are underway to confirm these results and determine if the R-CHOP + lenalidomide regimen is superior to R-CHOP in the up-front treatment of GBC and/or non-GBC variants.[14]
Treatment of recurrent and refractory DLBCL, NOS
Patients with DLBCL, NOS who relapse or progress following first-line therapy have been treated with "salvage regimens" consisting of high-dose (also termed high-intensity) chemotherapy conditioning drugs followed by autologous stem cell transplantation. This regimen has attained 3-year progression-free survival rates of 21–37%.[14] Relapse following this treatment carries a very poor prognosis with median overall survival times of ~10 months.[28] Patients who have failed or because of health issues are ineligible for autologous stem cell transplantation have been treated with low-dose (i.e. low-intensity) chemotherapy conditioning regimens followed by allogeneic stem cell transplantation. This regimen has achieved 3 year progression-free and overall survival rates of 41% and 52%, respectively.[32] Further studies are underway to determine the best treatment regimens for these cases.[14][32] Patients refractory to first-line therapy or who relapse within 12 months of receiving salvage therapy (including bone marrow transplant) for recurrent disease have had poor prognoses with median overall survival rates of 3.3 and 6.3 months, respectively.[14] The prognosis of these patients appears to be improved by using CAR-T therapy.
Chimeric antigen receptor T cell (i.e. CAR-T) adoptive cellular immunotherapy has emerged as a recent advance in treating refractory and relapsed DLBCL, NOS (tisagenlecleucel, axicabtagene ciloleucel, lisocabtagene maraleucel). Chimeric antigen receptor T cells are genetically engineered to express: 1) an artificial T-cell receptor consisting of antigen-recognition and attached hinge domains expressed on their surface membranes; 2) a surface membrane-spanning domain; 3) an intracellular domain which, when the antigen-recognition domain binds its targeted antigen, activates signaling pathways that cause the T-cell to attack and kill cells that bear the recognized antigen on their surface membranes; and 4), in more recently devised second generation CAR-T strategies, an associated intracellular co-stimulating molecule (e.g. CD28 or 4-1BB) which augments activation of the cell-killing signaling pathways. CAR-T therapy, as it pertains to DLBCL, NOS, kills a patient's neoplastic B-cells by isolating this patient's T-cells; genetically engineering these cells to express an artificial T-cell receptor designed to bind an antigen expressed on the surface of their neoplastic B-cells; and infusing these cells back into the donor patient. The targeted antigen has usually been CD19, a surface membrane protein expressed on virtually all B-cells including the neoplastic cells in DLBCL, NOS.[28] However, design of CARs[33] as well as the antigens chosen to be their targets [28] are constantly being changed in order to improve the efficacy of this therapeutic strategy.
CAR-T therapy for DLBCL, NOS has been used on patients who are refractory to and/or have progressed on first-line as well as salvage (including autologous stem cell transplantation) treatment regimens. Patients are treated first with a conditioning chemotherapy regimen, usually cyclophosphamide and fludarabine, and then infused with their own T-cells that have been engineered to attack CD19-bearing or, rarely, CD20-bearing cells. A meta-analysis of 17 studies using this or very similar approaches to treat DLBCL, NOS found the treatments gave complete and partial responses rates of 61% and 43%, respectively. While these studies did not have control groups and were too recent for meaningful estimates of remission durations, the remission rates were higher than expected using other treatment approaches. Significant and potentially lethal therapeutic complications of this therapy included development of the cytokine release syndrome (21% of cases), neurotoxicity, i.e. the CAR-T cell-related encephalopathy syndrome (9% of cases),[34] and the hemophagocytic lymphohistiocytosis/macrophage-activation syndrome (i.e. a form of Hemophagocytic lymphohistiocytosis).[35] Individual studies within and outside of this meta-analysis have reported remissions lasting >2 years but also lethal cytokine release syndrome and neurotoxicity responses to this therapy.[32] As a consequence of these studies, the Committee for Advanced Therapies and the Committee for Medicinal Products for Human Use of the European Medicines Agency recommend granting marketing authorization for tisagenlecleucel (i.e. chimeric antigen receptor T cells directed against CD19) in adult patients with DLBCL, NOS who have relapsed after or are refractory to two or more lines of systemic therapy.[36] The Committee for Orphan Medicinal Products of the European Medicines Agency recommends tisagenlecleucel retain its orphan drug designation.[36] The USA Food and Drug Administration (FDA) has also approved the use of this drug for relapsed or refractory DLBCL of the large B-cell lymphoma subtype in patients who have failed after two or more lines of systemic therapy.[37] Monoclonal antibodies directed against CD19, CD22, CD30, and PD-L1 have been developed for use as immunotherapeutic agents in other hematological malignancies and are being or plan to be tested for their usefulness in DLBCL, NOS.[22] In August 2020, the FDA approved the humanized Fc-modified cytolytic CD19 targeting monoclonal antibody tafasitamab in combination with lenalidomide as a treatment for adult patients with relapsed or refractory DLBCL.[38] In April 2021, the FDA approved the CD19-directed antibody-drug conjugate loncastuximab tesirine as a treatment for adult patients with relapsed or refractory DLBCL after systemic therapy.[39]
Emerging therapies
Neoplastic cell expression of CD30 in DLBCL, NOS is a favorable prognostic indicator; in these cases, brentuximab vedotin may be a useful addition to chemotherapy treatment protocols. This agent is a CD30-targeting antibody that delivers a toxin, monomethyl auristatin E, to CD30-expressing cells, has therapeutic efficacy against other CD30-expressing lymphomas, and may prove useful in treating the 10–15% of DLBCL, NOS cases expressing this protein. The neoplastic cells in the GBC variant of DLBCL, NOS often have mutations in the EZH2, BCL2 and CREBBP genes and overactive PI3K/AKT/mTOR and JAK-STAT signaling pathways while neoplastic cells in the ABC variant often have mutations in the MYD88, CD79A and CD79B (polatuzumab vedotin) genes and overactive B-cell receptor, toll-like receptor, and NF-κB signaling pathways.[22] These different gene mutations and dysregulated signaling pathways are also being studied as potential therapeutic targets for the individualized treatment of GBC and ABC/non-GBC cases.[14] CUDC-907, an inhibitor of PI3K and histone deacetylases, is being evaluated in two separate clinical trials[40][41] for the treatment of refractory and/or relapsed DLBCL, NOS including cases with alterations in the MYC gene.[42] GSK525762, an inhibitor of the BET family of proteins, suppresses expression of the MYC gene and is undergoing a phase I clinical trial[43] for the treatment of high-grade B-cell lymphoma with MYC, BL2, and/or BL6 rearrangements (i.e. DH/THL). RO6870810, another BET inhibitor, in combination with Venetoclax, an inhibitor of the Bcl-2 protein, is likewise in a phase I clinical trial[44] for the treatment of DH/THL.[42] Pharmacological inhibition of BCL-2 is effective in most B cell lymphomas, but often leads to acquired resistance due to the expression of other major anti-apoptotic BCL-2 family proteins like BCL-XL and MCL-1.[45] Combined therapy using MCL-1 inhibitor (S63845) or BCL-XL inhibitor (A-1331852) in addition to Venetoclax can be a solution to overcome this issue.[46]
Subtypes of diffuse large B-cell lymphoma
DLBCL subtypes have been sorted into groups based on their distinctive morphology or immunophenotype, distinctive clinical issues, and distinctive virus-driven etiology. The prognoses and treatment of these subtypes varies with their severity. Most subtypes are aggressive diseases and consequently treated in a manner similar to DLBCL,NOS. Further details on these subtypes, including their treatments, can be found in their respective main article linkages.
T cell/histiocyte-rich large B-cell lymphoma
T-cell/histiocyte-rich large B-cell lymphoma (THRLBCL) is a DLBCL in which tumors containing small numbers of usually large neoplastic B-cells embedded in a background of reactive T-cells and histiocytes develop in the liver, spleen, bone marrow and/or, rarely other sites. Patients usually present with advanced disease; their overall 3 year survival rates in different studies range between 46% and 72%.[12]
ALK+ large B-cell lymphoma
ALK+ large B-cell lymphoma (ALK+ LBCL) is a DLBCL in which neoplastic lymphocytes that express the ALK tyrosine kinase receptor protein infiltrate lymph nodes as well as extranodal sites, e.g. the mediastinum, bones, bone marrow, nasopharynx, tongue, stomach, liver, spleen, and skin. About 60% of these individuals present with advanced disease. ALK+ LBCL has an overall 5 year survival rate of ~34%.[12]
Plasmablastic lymphoma
Plasmablastic lymphoma (PBL) is a DLBCL in which neoplastic immunoblastic or plasmablastic cells embedded in a background of other cell types infiltrate the oral/nasal cavity or much less often the gastrointestinal tract.[12] Some 70% of individuals with PBL are infected with EBV[47] and/or (particularly those with oral/nasal cavity disease) human immunodeficiency virus (HIV).[12] PBL is an aggressive disease with a median survival time of ~15 months.[12]
Intravascular large B-cell lymphoma
Intravascular large B-cell lymphoma (IVLBCL) is a DLBCL in which medium- to large-sized neoplastic B-cells infiltrate small- to medium-sized blood vessels and sinusoids in the liver, spleen, and/or bone marrow. IVLBCL may be associated with the hemophagic syndrome (i.e. excessive cytokine secretion and systemic inflammation). Patients with the latter syndrome have very short survival times.[12] The poor prognosis of this disease has been significantly improved by rituximab or similar immunochemotherapy drugs but significant proportions of these responding cases relapse, often with central nervous system involvement.[48]
Large B-cell lymphoma with IRF4 rearrangement
Large B-cell lymphoma with IRF4 rearrangement (LBCL with IRF4 rearrangement) is a DLBCL in which tissue infiltrates containing intermediate- or large-sized neoplastic B-cells strongly express a chromosomal translocation involving the IRF4 gene on the short arm of chromosome 6. These cells form follicular, follicular and diffuse, or entirely diffuse infiltrates[12] in Waldeyer's tonsillar ring or other regions of the head and neck. The disease, which represents ~0.05% of all DLBCL, occurs primarily in children and young adults and typically has a good prognoses.[24] Cases with a follicular pattern of tissue infiltrates often have indolent disease and an excellent prognosis following excision and may not need chemotherapy. Cases with a purely diffuse tissue infiltrate pattern, in contrast, often do require chemotherapy.[12]
Primary mediastinal large B-cell lymphoma
Primary mediastinal large B-cell lymphoma (PMBL), also termed primary mediastinal (thymic) large B-cell lymphoma, is a DLBCL in which neoplastic B-cells infiltrates are commonly located in sclerotic/fibrous tissues of the thymus and mediastinal lymph nodes. The disease represents 6–10% of all DLBCL cases, presents with early stage disease in ~80% of cases, and has an overall survival rate at 5 years of 75–85%.[12]
Primary cutaneous diffuse large B-cell lymphoma, leg type
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBCL-LT) is a DLBCL in which diffuse patterns of immunoblastic and/or centroblastic B-cells infiltrate the dermis and/or subcutaneous tissue principally, but not exclusively, of the legs. This disease's 5-year overall survival rate is 50–60%.[12]
Primary diffuse large B-cell lymphoma of the central nervous system
Primary diffuse large B-cell lymphoma of the central nervous system (DLBCL-CNS, also termed primary central nervous system lymphoma [PCNSL]) is a DLBCL in which diffuse patterns of neoplastic B-cells with centroblastic, immunoblastic, or poorly differentiated features infiltrate the brain, spinal cord, leptomeninges, or eye.[24] The disease usually presents as a single lesion with a predilection for the supratentorial region of the brain but may involve the eye in 15–25% of cases, the cerebrospinal fluid in 7–42% of cases, and the spinal cord in ~1% of cases. The disease has a 5-year overall survival rate of ~30%.[12]
Diffuse large B-cell lymphoma associated with chronic inflammation
Diffuse large B-cell lymphoma associated with chronic inflammation (DLBCL-CI) is an Epstein–Barr virus-associated lymphoproliferative disease arising in persons with a long and persistent history of chronic inflammation. The disease's lesions consist of large, mature-appearing B-cells infiltrating the lung's pleura and nearby tissues. Most cases have occurred in patients who were given a pneumothorax (i.e. therapeutic introduction of air into the chest cavity in order to collapse and thereby "rest" the lung) to treat pulmonary tuberculosis that had progressed to a pyothorax (i.e. pus in the pleural cavity). Fibrin-associated large B-cell lymphoma (FA-DLBCL), often considered a sub-type of DLBCL-CI, is an infiltration of large neoplastic B-cells and fibrin affix to a prosthesis (e.g., cardiac valve, orthopaedic device) or accumulate within a hydrocele, pseudocyst, cardiac myxoma, or chronic subdural hematoma. The B-cells in these lesions are often but not always infected with the Epstein–Barr virus.[12] DLBCL-CI occurring in cases of pleural empyema (sometimes termed pyothorax-associated lymphoma, i.e. PAL) is an aggressive lymphoma with a five-year overall survival rate of 20–35%; FA-DLBCL, when involving the heart (e.g. occurring on myxommas or prosthetic valves) or vasculature structures (e.g. on thrombus-laden vascular grafts), may involve life-threatening cardiovascular complications, particularly strokes. Outside of these complications, however, DLBCL-CI usually has a highly favorable outcome.[24]
Lymphomatoid granulomatosis
Lymphomatoid granulomatosis (LYG) is a DLBCL in which large, atypical B-cells with immunoblastic or Hodgkin disease-like features that are infected by the Epstein-Barr virus center around and destroy the microvasculature. Lymphomatoid granulomatosis almost always involves the lung but may concurrently involve the brain, peripheral nervous system, skin, kidneys, liver, gastrointestinal tract, and/or upper respiratory tract; LYG has an increased incidence in persons with Wiskott–Aldrich syndrome or HIV or who are immunosuppression due to chemotherapy or organ transplantation.[12] The disease's prognosis is highly variable: patients with low grade disease often require no therapy except watchful waiting while patients with high grade disease usually require chemotherapy.[49]
Primary effusion lymphoma
Primary effusion lymphoma (PEL) is a DLBCL in which neoplastic B cells that resemble immunoblasts, plasmablasts, or Reed–Sternberg cells infiltrate the pleural, pericardial, or peritoneal membranes that surround the lungs, heart, and abdominal organs, respectively. This infiltration leads to the seeping of fluid into the cavities which are encased by these membranes, i.e. it leads to pleural effusions, pericardial effusions, and abdominal ascites. Some cases of PEL also involve the gastrointestinal tract and lymph nodes. The disease occurs primarily in people who are immunosuppressed or test positive for HIV[12] and are also latently infected with Kaposi's sarcoma-associated herpesvirus;[13] PEL is an aggressive disease with an overall 1 year survival rate of ~30%.[13]
Epstein–Barr virus-positive diffuse large B-cell lymphoma, not otherwise specified
Epstein–Barr virus-positive diffuse large B cell lymphoma, not otherwise specified (EBV+ DLBCL, NOS) is a B-cell lymphoma in which neoplastic B-cells that are infected with the Epstein-Barr virus cause a disease that does not fit into other subtypes of DLBCL. In EBV+ DLBCL, small neoplastic B-cells, other lymphocyte typess, plasma cells, histiocytes and epithelioid cells interspersed with Reed–Sternberg-like cells[24] infiltrate, almost exclusively, lymph nodes.[11] Elderly patients with the disease have median survival times of ~2 years while young patients have long-term treatment-related remissions in >80% of cases.[24]
HHV8-positive diffuse large B-cell lymphoma, NOS
HHV8-positive diffuse large B-cell lymphoma, NOS (HHV8+ DLBCL, NOS; also termed HHV8-positive diffuse large B-cell lymphoma [HHV8+ DLBCL]) is a DLBCL in which Kaposi's sarcoma-associated herpesvirus-infected, medium- to large-size neoplastic B-cells that resemble lymphocytes or immunoblasts infiltrate lymph nodes (~80% of cases) and, when disseminated (20% of cases), the liver and spleen. This infiltration usually disrupts the normal architecture of the involved tissues. HHV8+ DLBCL develops in HIV-infected individuals in ~50% of cases, in individuals with multicentric Castleman disease, plasma cell variant in uncommon cases, and in individuals with Kaposi sarcoma in rare cases. HHV8+ DLBCL commonly takes an aggressive course and has a poor prognosis.[12]
Related disorders
Helicobactor pylori associated diffuse large B-cell lymphoma
Rare cases of DLBCL are associated with the presence of the bacterium, Helicobacter pylori, in the neoplastic B-cells.[7] While the histology of Helicobactor pylori-associated diffuse large B-cell lymphoma (H. pylori+ DLBCL) is typical of DLBCL, the disease is sometimes a progression of mantle cell lymphoma, is often restricted to the stomach, is less aggressive that most DLBCL cases, and may respond to a drug regimen consisting of antibiotics and proton pump inhibitors directed at killing the bacterium.[50][23] Perhaps because of these features of the disease, H. pylori+ DLBCL has not been classified as a DLBCL by the World Health Organization, 2016.[23]
Recent studies suggest that localized, early-stage H. pylori+ DLBCL, when limited to the stomach, is successfully treated with H. pylori eradication protocols consisting of two or more antibiotics plus a proton pump inhibitor.[51][50][52][23] However, these studies also agree that patients treated with one of these H. pylori eradication regimes need to be carefully followed: those unresponsive to, or worsening on, these regimens should be switched to a chemotherapy regimen (e.g. R-CHOP) and/or, for complicated bulky disease, surgery and/or local radiotherapy.[50][23]
Epstein–Barr virus-positive mucocutaneous ulcer
Epstein-Barr virus-positive mucocutaneous ulcer (EBVMCU) was first described as a lymphoproliferative disorder in which Epstein–Barr virus-infected B-cells proliferate and cause ulcerations in the mucous membranes and skin of immunosuppressed individuals. Its lesions consist of Epstein–Barr virus-positive, variable-sized, atypical B-cells that by conventional histopathologic criteria indicate the lesions are a form of DLBCL. Since these lesions regress spontaneously without anti-cancer treatment, EBVMCU is now considered a pseudo-malignant disorder.[53] Elderly individuals that evidence the disease but have no other cause for immunosuppression may exhibit a relapsing and remitting course with their ulcers worsening but then regressing spontaneously.[54] Persistent and/or severely symptomatic cases have had excellent responses to rituximab.[55] Individuals developing these ulcers as a consequence of immunosuppressive therapy generally have a remission after the dosage of the drugs used in their immunosuppressive treatments are reduced. Most of these patients do not relapse.[54]
References
- "A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. The Non-Hodgkin's Lymphoma Classification Project". Blood. 89 (11): 3909–3918. June 1997. doi:10.1182/blood.V89.11.3909. PMID 9166827.
- Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS (January 2006). "Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001". Blood. 107 (1): 265–276. doi:10.1182/blood-2005-06-2508. PMC 1895348. PMID 16150940.
- Smith A, Howell D, Patmore R, Jack A, Roman E (November 2011). "Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network". British Journal of Cancer. 105 (11): 1684–1692. doi:10.1038/bjc.2011.450. PMC 3242607. PMID 22045184.
- Smith A, Roman E, Howell D, Jones R, Patmore R, Jack A (March 2010). "The Haematological Malignancy Research Network (HMRN): a new information strategy for population based epidemiology and health service research". British Journal of Haematology. 148 (5): 739–753. doi:10.1111/j.1365-2141.2009.08010.x. PMC 3066245. PMID 19958356.
- Kumar V, Abbas AK, Fausto N, Aster JC (28 May 2009). Robbins & Cotran Pathologic Basis of Disease. Elsevier Health Sciences. p. 607. ISBN 978-1-4377-2015-0.
- Freeman AS, Aster JS (2012). "Epidemiology, clinical manifestations, pathologic features, and diagnosis of diffuse large B cell lymphoma". In Basow DS (ed.). UpToDate. Waltham, MA: UpToDate.
- Casulo C, Friedberg J (2017). "Transformation of marginal zone lymphoma (and association with other lymphomas)". Best Practice & Research. Clinical Haematology. 30 (1–2): 131–138. doi:10.1016/j.beha.2016.08.029. PMID 28288708.
- Abuelgasim KA, Rehan H, Alsubaie M, Al Atwi N, Al Balwi M, Alshieban S, Almughairi A (March 2018). "Coexistence of chronic myeloid leukemia and diffuse large B-cell lymphoma with antecedent chronic lymphocytic leukemia: a case report and review of the literature". Journal of Medical Case Reports. 12 (1): 64. doi:10.1186/s13256-018-1612-4. PMC 5845776. PMID 29524963.
- Swerdlow et al. 2008, p. 233.
- Castillo JJ, Beltran BE, Miranda RN, Young KH, Chavez JC, Sotomayor EM (July 2018). "EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2018 update on diagnosis, risk-stratification and management". American Journal of Hematology. 93 (7): 953–962. doi:10.1002/ajh.25112. PMID 29984868.
- Nicolae A, Pittaluga S, Abdullah S, Steinberg SM, Pham TA, Davies-Hill T, Xi L, Raffeld M, Jaffe ES (August 2015). "EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment". Blood. 126 (7): 863–72. doi:10.1182/blood-2015-02-630632. PMC 4536540. PMID 25999451.
- Sukswai N, Lyapichev K, Khoury JD, Medeiros LJ (November 2019). "Diffuse large B-cell lymphoma variants: an update". Pathology. 52 (1): 53–67. doi:10.1016/j.pathol.2019.08.013. PMID 31735345.
- Shimada K, Hayakawa F, Kiyoi H (November 2018). "Biology and management of primary effusion lymphoma". Blood. 132 (18): 1879–1888. doi:10.1182/blood-2018-03-791426. PMID 30154110.
- Liu Y, Barta SK (May 2019). "Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment". American Journal of Hematology. 94 (5): 604–616. doi:10.1002/ajh.25460. PMID 30859597.
- Goldman & Schafer 2012, p. 1222.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (May 2016). "The 2016 revision of the World Health Organization classification of lymphoid neoplasms". Blood. 127 (20): 2375–90. doi:10.1182/blood-2016-01-643569. PMC 4874220. PMID 26980727.
- Goldman & Schafer 2012, p. 1225.
- Akyurek N, Uner A, Benekli M, Barista I (September 2012). "Prognostic significance of MYC, BCL2, and BCL6 rearrangements in patients with diffuse large B-cell lymphoma treated with cyclophosphamide, doxorubicin, vincristine, and prednisone plus rituximab". Cancer. 118 (17): 4173–4183. doi:10.1002/cncr.27396. PMID 22213394. S2CID 19134744.
- Feugier P, Van Hoof A, Sebban C, Solal-Celigny P, Bouabdallah R, Fermé C, et al. (June 2005). "Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d'Etude des Lymphomes de l'Adulte". Journal of Clinical Oncology. 23 (18): 4117–4126. doi:10.1200/JCO.2005.09.131. PMID 15867204. S2CID 23556248.
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. (February 2000). "Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling". Nature. 403 (6769): 503–511. Bibcode:2000Natur.403..503A. doi:10.1038/35000501. PMID 10676951. S2CID 4382833.
- Swerdlow et al. 2008, pp. 233–7.
- Li S, Young KH, Medeiros LJ (January 2018). "Diffuse large B-cell lymphoma". Pathology. 50 (1): 74–87. doi:10.1016/j.pathol.2017.09.006. PMID 29167021. S2CID 20839613.
- Cheng Y, Xiao Y, Zhou R, Liao Y, Zhou J, Ma X (August 2019). "Prognostic significance of helicobacter pylori-infection in gastric diffuse large B-cell lymphoma". BMC Cancer. 19 (1): 842. doi:10.1186/s12885-019-6067-5. PMC 6712724. PMID 31455250.
- Grimm KE, O'Malley DP (February 2019). "Aggressive B cell lymphomas in the 2017 revised WHO classification of tumors of hematopoietic and lymphoid tissues". Annals of Diagnostic Pathology. 38: 6–10. doi:10.1016/j.anndiagpath.2018.09.014. PMID 30380402. S2CID 53196244.
- Ollila TA, Olszewski AJ (June 2018). "Extranodal Diffuse Large B Cell Lymphoma: Molecular Features, Prognosis, and Risk of Central Nervous System Recurrence". Current Treatment Options in Oncology. 19 (8): 38. doi:10.1007/s11864-018-0555-8. PMC 6294323. PMID 29931605.
- Pileri S, Ponzoni M (2017). "Pathology of nodal marginal zone lymphomas". Best Practice & Research. Clinical Haematology. 30 (1–2): 50–55. doi:10.1016/j.beha.2016.11.001. PMID 28288717.
- Abramson JS (September 2019). "Hitting back at lymphoma: How do modern diagnostics identify high-risk diffuse large B-cell lymphoma subsets and alter treatment?". Cancer. 125 (18): 3111–3120. doi:10.1002/cncr.32145. PMID 31287161.
- Chavez JC, Locke FL (June 2018). "CAR T cell therapy for B-cell lymphomas". Best Practice & Research. Clinical Haematology. 31 (2): 135–146. doi:10.1016/j.beha.2018.04.001. PMC 6716161. PMID 29909914.
- Shahjahani M, Norozi F, Ahmadzadeh A, Shahrabi S, Tavakoli F, Asnafi AA, Saki N (January 2015). "The role of Pax5 in leukemia: diagnosis and prognosis significance". Medical Oncology (Northwood, London, England). 32 (1): 360. doi:10.1007/s12032-014-0360-6. PMID 25428382. S2CID 7127158.
- Cabanillas F, Shah B (December 2017). "Advances in Diagnosis and Management of Diffuse Large B-cell Lymphoma". Clinical Lymphoma, Myeloma & Leukemia. 17 (12): 783–796. doi:10.1016/j.clml.2017.10.007. PMID 29126866. S2CID 25304758.
- Twa DDW, Mottok A, Savage KJ, Steidl C (May 2018). "The pathobiology of primary testicular diffuse large B-cell lymphoma: Implications for novel therapies". Blood Reviews. 32 (3): 249–255. doi:10.1016/j.blre.2017.12.001. PMID 29289361.
- Gisselbrecht C, Van Den Neste E (September 2018). "How I manage patients with relapsed/refractory diffuse large B cell lymphoma". British Journal of Haematology. 182 (5): 633–643. doi:10.1111/bjh.15412. PMC 6175435. PMID 29808921.
- Lee YH, Kim CH (July 2019). "Evolution of chimeric antigen receptor (CAR) T cell therapy: current status and future perspectives". Archives of Pharmacal Research. 42 (7): 607–616. doi:10.1007/s12272-019-01136-x. PMID 30830661. S2CID 73475110.
- Zheng XH, Zhang XY, Dong QQ, Chen F, Yang SB, Li WB (January 2020). "Efficacy and safety of chimeric antigen receptor-T cells in the treatment of B cell lymphoma: a systematic review and meta-analysis". Chinese Medical Journal. 133 (1): 74–85. doi:10.1097/CM9.0000000000000568. PMC 7028209. PMID 31923107.
- Hopfinger G, Jäger U, Worel N (April 2019). "CAR-T Cell Therapy in Diffuse Large B Cell Lymphoma: Hype and Hope". HemaSphere. 3 (2): e185. doi:10.1097/HS9.0000000000000185. PMC 6746029. PMID 31723824.
- Ali S, Kjeken R, Niederlaender C, Markey G, Saunders TS, Opsata M, Moltu K, Bremnes B, Grønevik E, Muusse M, Håkonsen GD, Skibeli V, Kalland ME, Wang I, Buajordet I, Urbaniak A, Johnston J, Rantell K, Kerwash E, Schuessler-Lenz M, Salmonson T, Bergh J, Gisselbrecht C, Tzogani K, Papadouli I, Pignatti F (October 2019). "The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma". The Oncologist. 25 (2): theoncologist.2019–0233. doi:10.1634/theoncologist.2019-0233. PMC 7011647. PMID 31619548.
- Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, Blumenthal GM, Bryan W, McKee AE, Pazdur R (March 2019). "FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma". Clinical Cancer Research. 25 (6): 1702–1708. doi:10.1158/1078-0432.CCR-18-2743. PMID 30413526.
- "Monjuvi: FDA Approved Drugs". US FDA. Retrieved August 20, 2020.
- "FDA grants accelerated approval to loncastuximab tesirine-lpyl for large B-cell lymphoma". FDA. 11 June 2021.
- "Study to Evaluate the Efficacy and Safety of CUDC-907 in Patients With RR DLBCL, Including Patients With MYC Alterations". ClinicalTrials.gov. 30 August 2019.
- "Study to Assess the Safety, Tolerability and Pharmacokinetics of Fimepinostat (CUDC-907) in Patients With Lymphoma". ClinicalTrials.gov. 5 May 2021.
- Ok CY, Medeiros LJ (January 2020). "High-grade B-cell lymphoma: a term re-purposed in the revised WHO classification". Pathology. 52 (1): 68–77. doi:10.1016/j.pathol.2019.09.008. PMID 31735344.
- "Testing a New Anti-cancer Drug Combination, Entinostat and GSK525762C, for Advanced and Refractory Solid Tumors and Lymphomas". ClinicalTrials.gov. 25 September 2020.
- "A Study to Evaluate Safety, Pharmacokinetics, and Clinical Activity of Combination of RO6870810 and Venetoclax, With or Without Rituximab, in Participants With Relapsed/Refractory DLBCL and/or High-Grade B-Cell Lymphoma and/or High Grade B-Cell Lymphoma With MYC and/or BCL2 and/or BCL6". ClinicalTrials.gov. 27 January 2021.
- Zhao X, Ren Y, Lawlor M, Shah BD, Park PM, Lwin T, et al. (May 2019). "BCL2 Amplicon Loss and Transcriptional Remodeling Drives ABT-199 Resistance in B Cell Lymphoma Models". Cancer Cell. 35 (5): 752–766.e9. doi:10.1016/j.ccell.2019.04.005. PMC 6945775. PMID 31085176.
- Dengler MA, Teh CE, Thijssen R, Gangoda L, Lan P, Herold MJ, et al. (February 2020). "Potent efficacy of MCL-1 inhibitor-based therapies in preclinical models of mantle cell lymphoma". Oncogene. 39 (9): 2009–2023. doi:10.1038/s41388-019-1122-x. PMID 31772331. S2CID 208302779.
- Rezk SA, Zhao X, Weiss LM (June 2018). "Epstein—Barr virus-associated lymphoid proliferations, a 2018 update". Human Pathology. 79: 18–41. doi:10.1016/j.humpath.2018.05.020. PMID 29885408. S2CID 47010934.
- Ponzoni M, Campo E, Nakamura S (October 2018). "Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks". Blood. 132 (15): 1561–1567. doi:10.1182/blood-2017-04-737445. PMID 30111607.
- Chavez JC, Sandoval-Sus J, Horna P, Dalia S, Bello C, Chevernick P, Sotomayor EM, Sokol L, Shah B (August 2016). "Lymphomatoid Granulomatosis: A Single Institution Experience and Review of the Literature". Clinical Lymphoma, Myeloma & Leukemia. 16 (Suppl): S170–4. doi:10.1016/j.clml.2016.02.024. PMID 27521314.
- Kuo SH, Yeh KH, Chen LT, Lin CW, Hsu PN, Hsu C, Wu MS, Tzeng YS, Tsai HJ, Wang HP, Cheng AL (June 2014). "Helicobacter pylori-related diffuse large B-cell lymphoma of the stomach: a distinct entity with lower aggressiveness and higher chemosensitivity". Blood Cancer Journal. 4 (6): e220. doi:10.1038/bcj.2014.40. PMC 4080211. PMID 24949857.
- Paydas S (April 2015). "Helicobacter pylori eradication in gastric diffuse large B cell lymphoma". World Journal of Gastroenterology. 21 (13): 3773–6. doi:10.3748/wjg.v21.i13.3773. PMC 4385524. PMID 25852262.
- Tsai HJ, Tai JJ, Chen LT, Wu MS, Yeh KH, Lin CW, Wang TE, Wang HP, Yu FJ, Liou JM, Hsiao CF, Cheng TY, Yeh HJ, Ko CW, Chen MJ, Lo GH, Hsu PI, Chang CS, Hwang WS, Chuang SS, Lee HW, Shun CT, Chiu CF, Wang WM, Hsieh CY, Liu TW, Lin JT, Kuo SH, Cheng AL (November 2019). "A multicenter prospective study of first-line antibiotic therapy for early-stage gastric mucosa-associated lymphoid tissue lymphoma and diffuse large B-cell lymphoma with histological evidence of mucosa-associated lymphoid tissue". Haematologica. 105 (7): e349–e354. doi:10.3324/haematol.2019.228775. PMC 7327622. PMID 31727764.
- Ikeda T, Gion Y, Yoshino T, Sato Y (2019). "A review of EBV-positive mucocutaneous ulcers focusing on clinical and pathological aspects". Journal of Clinical and Experimental Hematopathology. 59 (2): 64–71. doi:10.3960/jslrt.18039. PMC 6661964. PMID 31257347.
- Goodlad JR (June 2017). "Epstein–Barr Virus-associated Lymphoproliferative Disorders in the Skin". Surgical Pathology Clinics. 10 (2): 429–453. doi:10.1016/j.path.2017.01.001. PMID 28477890.
- Dojcinov SD, Fend F, Quintanilla-Martinez L (March 2018). "EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts". Pathogens (Basel, Switzerland). 7 (1): 28. doi:10.3390/pathogens7010028. PMC 5874754. PMID 29518976.
Sources
- Swerdlow SH, Campo E, Jaffe ES, Pileri SA, eds. (2008). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC. ISBN 978-92-832-2431-0.
- Goldman L, Schafer AI (2012). Goldman's Cecil Medicine (24th ed.). ISBN 978-1-4377-1604-7.
- Turgeon ML (2005). Clinical hematology: theory and procedures. Hagerstown, MD: Lippincott Williams & Wilkins. ISBN 978-0-7817-5007-3.