Kjer's optic neuropathy
| Kjer's optic neuropathy | |
|---|---|
| Other names: Autosomal dominant optic atrophy, Kjer type; Kjer optic atrophy; or, Kjer's autosomal dominant optic atrophy. | |
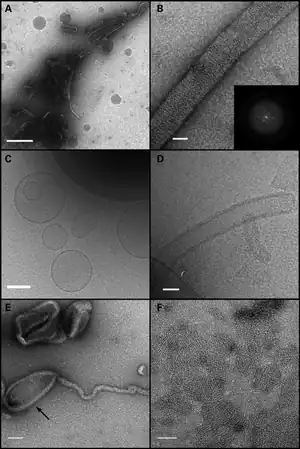 | |
| Electron microscopy of liposome tubulation. (A and B) Negative stain of OPA1-S1 reconstituted with liposomes mimicking the mitochondrial inner membrane lipid. (A) A low magnification view showing OPA1-S1 tubular structures with diameters in the range of 50 to 150 nm. In (B), a higher magnification view shows an individual membrane tubule with a periodic arrangement of OPA1-S1 on the lipid substrate. The inset represents the Fourier transform of a segment of the displayed tube indicating molecular order mainly along the long axis of the tube. (C) Cryo-TEM image of vesicle preparations prior to the addition of OPA1-S1. Frozen–hydrated vesicles appear as a mixture of unilamellar and multilamellar vesicles of varying diameters. (D) Cryo-TEM of the vesicle preparation after the addition of OPA1-S1. OPA1-S1 reconstitutes into tubular structures with OPA1-S1 molecules decorating the surface of the lipid tubules. (E) Negative stain of OPA1 mixed with liposomes, showing that OPA1 assembles on the tubular as well as non-tubular (arrow) portions of the liposome. (F) Negative-stained preparation of an OPA1/liposome reaction, showing patches of OPA1 arrays. Scale bars: (A) 500 nm; (B and D) 50 nm; (C) 200 nm; (E and F) 100 nm. | |
Dominant optic atrophy, or dominant optic atrophy, Kjer's type, is an autosomally inherited disease that affects the optic nerves, causing reduced visual acuity and blindness beginning in childhood. This condition is due to mitochondrial dysfunction mediating the death of optic nerve fibers. Dominant optic atrophy was first described clinically by Batten in 1896 and named Kjer’s optic neuropathy in 1959 after Danish ophthalmologist Poul Kjer, who studied 19 families with the disease.[1] Although dominant optic atrophy is the most common autosomally inherited optic neuropathy (i.e., disease of the optic nerves) aside from glaucoma, it is often misdiagnosed.
Signs and symptoms
Autosomal dominant optic atrophy can present clinically as an isolated bilateral optic neuropathy (non-syndromic form) or rather as a complicated phenotype with extra-ocular signs (syndromic form). Dominant optic atrophy usually affects both eyes roughly symmetrically in a slowly progressive pattern of vision loss beginning in childhood and is hence a contributor to childhood blindness. Vision testing will reveal scotomas (areas of impaired visual acuity) in the central visual fields with peripheral vision sparing and impaired color vision (color blindness). Visual acuity loss varies from mild to severe, typically ranging from 6/6 (in meters, equivalent to 20/20, ft) to 6/60 (20/200, ft) with a median value of 6/36 (roughly equivalent to 20/125 ft), corrected vision. In rare cases, vision loss is more severe.
Characteristic changes of the fundus evident on examination is temporal pallor (indicating atrophy) of the optic disc and in its end stage, excavation of the optic disc, as is also seen in Leber hereditary optic neuropathy and normal tension glaucoma.
Because the onset of Dominant optic atrophy is insidious, symptoms are often not noticed by the patients in its early stages and are picked up by chance in routine school eye screenings. First signs of Kjer's typically present between 4–6 years of age, though presentation at as early as 1 year of age has been reported. In some cases, Dominant optic atrophy may remain subclinical until early adulthood.
Progression of dominant optic atrophy varies even within the same family. Some have mild cases with visual acuity stabilizing in adolescence, others have slowly but constantly progressing cases, and others still have sudden step-like decreases in visual acuity. Generally, the severity of the condition by adolescence reflects the overall level of visual function to be expected throughout most of the patient’s adult life (Votruba, 1998). Slow decline in acuity is known to occur in late middle age in some families.
In complicated cases of autosomal dominant optic atrophy, in addition to bilateral optic neuropathy, several other neurological signs of neurological involvement can be observed: peripheral neuropathy, deafness, cerebellar ataxia, spastic paraparesis, myopathy.[2]
Genetics
Dominant optic atrophy is inherited in an autosomal dominant manner. That is, a heterozygous patient with the disease has a 50% chance of passing on the disease to offspring, assuming his/her partner does not have the disease. Males and females are affected at the same rate. Although Kjer's has a high penetrance (98%), severity and progression of DOA are extremely variable even within the same family.
Pathophysiology
Vision loss in dominant optic atrophy is due to optic nerve fiber loss from mitochondria dysfunction. Dominant optic atrophy is associated with mutation of the OPA1 gene[3] found on chromosome 3, region q28-qter. Also, 5 other chromosomal genes are described as causing optic atrophy: OPA2 (x-linked), OPA3 (dominant), OPA4 (dominant), OPA5 (dominant) and OPA6 (recessive) (see OMIM 165500).
The OPA1 gene codes for a dynamin-related GTPase protein targeted to the mitochondrial inner membrane. OPA1 has distinct roles in the fusion of mitochondrial inner membranes during mitochondrial fusion events, and in regulation of cell death.[4]
Mitochondria are subcellular structures that generate and transform energy from metabolism into discrete usable units (ATP) for the cell’s functions (See oxidative phosphorylation, electron transport chain). Retinal ganglion cells (neurons), which make up the optic nerve, have a high energy demand and are particularly sensitive to mitochondrial dysfunction. This is especially the case for smaller and less myelinated neurons such as those found in the papillomacular bundle of the retina, which transmit information corresponding to the central visual field. Biochemical and mitochondrial morphological studies on cells from patients affected by autosomal dominant optic atrophy have shown a severe defect in the shape (with a very remarkable fragmentation of the mitochondrial tubules in small spheres) and distribution of mitochondria, occurring independently from a bioenergetic defect (respiratory chain function, ATP synthesis, and reactive oxygen species production) or apoptosis, indicating that the mitochondrial fusion defect is the primary pathogenetic mechanism,[5] although variable bioenergetic defects can also occur as a secondary phenomenon, especially in severe cases with complicated phenotypes and accumulation of multiple mitochondrial-DNA deletions.
Over 60 different mutations of the OPA1 gene causing Kjer's have been reported, most of which occur in the catalytic domain of the protein.
Mutations at the OPA1 gene are also associated with normal tension glaucoma (OMIM 606657) and deafness (OMIM 125250).
Diagnosis
The evaluation of this condition is done via clinical (ocular) presentation, if the individual has tritanopia, family history, and can be confirmed with genetic testing [6]
Management
Currently there is no effective therapy for dominant optic atrophy, and consequently, these patients are simply monitored for changes in vision by their eye-care professional. Children of patients should be screened regularly for visual changes related to dominant optic atrophy. Research is underway to further characterize the disease so that therapies may be developed.
Since November 2018, Cure ADOA Foundation Archived 2021-09-09 at the Wayback Machine has been focusing on fellow patients and their families. They have the following goals: scientific research, disease awareness, interaction between all parties involved and a trustworthy place for the patients.
Incidence
The incidence of dominant optic atrophy has been estimated to be 1:50000 with prevalence as high as 1:10000 in the Danish population (Votruba, 1998).
See also
References
- ↑ Kjer, P (1959). "Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families". Acta Ophthalmologica Supplementum. 164 (Supp 54): 1–147. PMID 13660776.
- ↑ Yu-Wai-Man, P; Griffiths, PG; Gorman, GS; Lourenco, CM; Wright, AF; Auer-Grumbach, M; Toscano, A; Musumeci, O; Valentino, ML; Caporali, L; Lamperti, C; Tallaksen, CM; Duffey, P; Miller, J; Whittaker, RG; Baker, MR; Jackson, MJ; Clarke, MP; Dhillon, B; Czermin, B; Stewart, JD; Hudson, G; Reynier, P; Bonneau, D; Marques, W Jr; Lenaers, G; McFarland, R; Taylor, RW; Turnbull, DM; Votruba, M; Zeviani, M; Carelli, V; Bindoff, LA; Horvath, R; Amati-Bonneau, P; Chinnery, PF (March 2010). "Multi-system neurological disease is common in patients with OPA1 mutations". Brain : A Journal of Neurology. 133 (Pt 3): 771–86. doi:10.1093/brain/awq007. PMC 2842512. PMID 20157015.
- ↑ Delettre, C; Lenaers, G; Griffoin, JM; Gigarel, N; Lorenzo, C; Belenguer, P; Pelloquin, L; Grosgeorge, J; Turc-Carel, C; Perret, E; Astarie-Dequeker, C; Lasquellec, L; Arnaud, B; Ducommun, B; Kaplan, J; Hamel, CP (October 2000). "Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy". Nature Genetics. 26 (2): 207–10. doi:10.1038/79936. PMID 11017079.
- ↑ Frezza, C; Cipolat, S; Martins de Brito, O; Micaroni, M; Beznoussenko, GV; Rudka, T; Bartoli, D; Polishuck, RS; Danial, NN; De Strooper, B; Scorrano, L (Jul 14, 2006). "OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion". Cell. 126 (1): 177–89. doi:10.1016/j.cell.2006.06.025. PMID 16839885.
- ↑ Spinazzi, M; Cazzola, S; Bortolozzi, M; Baracca, A; Loro, E; Casarin, A; Solaini, G; Sgarbi, G; Casalena, G; Cenacchi, G; Malena, A; Frezza, C; Carrara, F; Angelini, C; Scorrano, L; Salviati, L; Vergani, L (Nov 1, 2008). "A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function". Human Molecular Genetics. 17 (21): 3291–302. doi:10.1093/hmg/ddn225. PMID 18678599.
- ↑ "Optic atrophy 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 29 August 2021. Retrieved 8 September 2021.
Further reading
- Carelli; Ross-Cisneros, FN; Sadun, AA (2004). "Mitochondrial dysfunction as a cause of optic neuropathies". Progress in Retinal and Eye Research. 23 (1): 53–89. doi:10.1016/j.preteyeres.2003.10.003. PMID 14766317.
- Entrez Gene OPA1 4976 Archived 2010-03-07 at the Wayback Machine
- OMIM: OPA1 deafness OMIM 125250 Archived 2021-03-27 at the Wayback Machine
- OMIM: OPA1 Normotension glaucoma OMIM 606657 Archived 2020-07-29 at the Wayback Machine
- OMIM: OPA1 OMIM 605290 Archived 2021-03-26 at the Wayback Machine
- OMIM: Optic Atrophy 1 OMIM 165500 Archived 2021-03-20 at the Wayback Machine
- Votruba; Moore, AT; Bhattacharya, SS (1998). "Clinical features, molecular genetics, and pathophysiology of dominant optic atrophy". Journal of Medical Genetics. 35 (10): 793–800. doi:10.1136/jmg.35.10.793. PMC 1051452. PMID 9783700.
External links
| Classification |
|---|