Axenfeld–Rieger syndrome
| Axenfeld–Rieger syndrome | |
|---|---|
| Other names: Axenfeld syndrome, Hagedoom syndrome | |
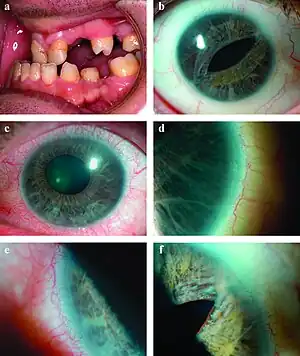 | |
| a) Microdontia and hypodontia. b) Slit pupil and iris atrophy right eye. c) Corectopia with iris atrophy left eye. d) Posterior embryotoxon right eye. e) Posterior embryotoxon left eye. f) Broad peripheral anterior synechiae right eye.[1] | |
Axenfeld–Rieger syndrome is a rare autosomal dominant[2] disorder, which affects the development of the teeth, eyes, and abdominal region.[3]
Signs and symptoms
.png.webp)
The clinical presentation of this condition is consistent with the following:[4]
- Everted lower lip vermilion
- Glaucoma
- Hearing impairment
- Midface retrusion
- Aplasia of the iris
- Hypertelorism
- Hypodontia
- Hypoplasia of maxilla
- Microdontia
- Inguinal hernia
- Zygomatic flattening
- Big lower jaw
- Megalocornea
Pathophysiology
The molecular genetics of Axenfeld–Rieger syndrome are poorly understood, but center on three genes identified by cloning of chromosomal breakpoints from patients.[6][7]
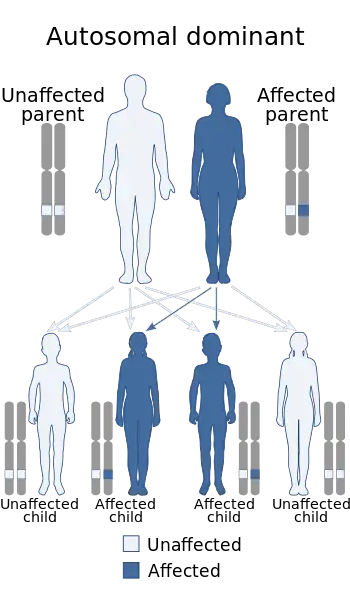
This disorder is inheritable as an autosomal dominant trait,[5] which means the defective gene is located on an autosome, and only one copy of the gene is sufficient to cause the disorder when inherited from a parent who has the disorder. As shown in the diagram, this gives a 50/50 chance of offspring inheriting the condition from an affected parent.
Diagnosis
Although most recognized for its correlation with the onset of glaucoma, the malformation is not limited to the eye, as Axenfeld–Rieger syndrome when associated with the PITX2 genetic mutation usually presents congenital malformations of the face, teeth, and skeletal system.[8]
The most characteristic feature affecting the eye is a distinct corneal posterior arcuate ring, known as an "embryotoxon".[9] In severe cases, iris may be adherent to the cornea anterior to the Schwalbe's line.[10]
One of the three known genetic mutations which cause Rieger syndrome can be identified through genetic samples analysis. About 40% of Axenfeld–Rieger sufferers have displayed mutations in genes PITX2,[8] FOXC1, and PAX6.[11][12] The difference between Type 1, 2, and 3 Axenfeld–Rieger syndrome is the genetic cause, all three types display the same symptoms and abnormalities.[13]
Classification
The OMIM classification is as follows:
| Type | OMIM | Gene |
|---|---|---|
| Type 1 | 180500 | PITX2 |
| Type 2 | 601499 | possibly FOXO1A[14] |
| Type 3 | 602482 | FOXC1 |
| DeHauwere syndrome | 109120 | Unknown[11] |
Detection of any of these mutations can give patients a clear diagnosis and prenatal procedures such as preimplantation genetic diagnosis, chorionic villus sampling and amniocentesis can be offered to patients and prospective parents.[15]
Management
Treatment for Axenfeld–Rieger syndrome should be based on clinical presentation, therefore a decrease intra-ocular pressure if there is glaucoma (Latanoprost or other). Should the individual have additional findings then surgery may be used to correct facial/dental problems, or cardiac issues.[4]
Eponym
It is named after the German ophthalmologist Theodor Axenfeld[16][9] who studied anterior segment disorders, especially those such as Rieger syndrome and the Axenfeld anomaly.
Axenfeld–Rieger syndrome is characterized by abnormalities of the eyes, teeth, and facial structure.[13] Rieger syndrome, by medical definition, is determined by the presence of malformed teeth, underdeveloped anterior segment of the eyes, and cardiac problems associated with the Axenfeld anomaly.[14] The term "Rieger syndrome" is sometimes used to indicate an association with glaucoma.[8] Glaucoma occurs in up to 50% of patients with Rieger syndrome. Glaucoma develops during adolescence or late childhood, but often occurs in infancy.[9][11] In addition, a prominent Schwalbe's line, an opaque ring around the cornea known as posterior embryotoxon, may arise with hypoplasia of the iris.[6] Below average height and stature, stunted development of the mid-facial features and mental deficiencies may also be observed in patients.[6]
See also
- Primary juvenile glaucoma
- SHORT syndrome
- Jade Etherington: Paralympic athlete
References
- ↑ Dhir, L; Frimpong-Ansah, K; Habib, Nabil E (2008). "Missed case of Axenfeld-Rieger syndrome: a case report". Cases Journal. 1 (1): 299. doi:10.1186/1757-1626-1-299. PMC 2585579. PMID 18990239.
- ↑ Vieira, Véronique; David, Gabriel; Roche, Olivier; de la Houssaye, Guillaume; Boutboul, Sandrine; Arbogast, Laurence; Kobetz, Alexandra; Orssaud, Christophe; Camand, Olivier; Schorderet, Daniel F.; Munier, Francis; Rossi, Annick; Delezoide, Anne Lise; Marsac, Cécile; Ricquier, Daniel; Dufier, Jean-Louis; Menasche, Maurice; Abitbol, M. (2006). "Identification of four new PITX2 gene mutations in patients with Axenfeld-Rieger syndrome". Molecular Vision. 12: 1448–60. PMID 17167399. Archived from the original on 2020-07-14. Retrieved 2021-09-19.
- ↑ Fitch, Naomi; Kaback, Martin (1978). "The Axenfeld syndrome and the Rieger syndrome". Journal of Medical Genetics. 15 (1): 30–4. doi:10.1136/jmg.15.1.30. PMC 1012820. PMID 416212.
- 1 2 "Axenfeld-Rieger syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 18 March 2021. Retrieved 24 September 2021.
- 1 2 "Axenfeld-Rieger syndrome type 1". National Center for Biotechnology Information. Archived from the original on 2020-11-04. Retrieved 2021-09-19.
- 1 2 3 Suzuki, Katsuhiro; Nakamura, Makoto; Amano, Emi; Mokuno, Kumiko; Shirai, Shoichiro; Terasaki, Hiroko (2006). "Case of chromosome 6p25 terminal deletion associated with Axenfeld–Rieger syndrome and persistent hyperplastic primary vitreous". American Journal of Medical Genetics Part A. 140 (5): 503–8. doi:10.1002/ajmg.a.31085. PMID 16470791.
- ↑ Tonoki, Hidefumi; Harada, Naoki; Shimokawa, Osamu; Yosozumi, Ayako; Monzaki, Kadomi; Satoh, Kohei; Kosaki, Rika; Sato, Atsushi; Matsumoto, Naomichi; Iizuka, Susumu (2011). "Axenfeld-Rieger anomaly and Axenfeld-Rieger syndrome: Clinical, molecular-cytogenetic, and DNA array analyses of three patients with chromosomal defects at 6p25". American Journal of Medical Genetics Part A. 155A (12): 2925–32. doi:10.1002/ajmg.a.33858. PMID 22009788.
- 1 2 3 Meyer-Marcotty, P.; Weisschuh, N.; Dressler, P.; Hartmann, J.; Stellzig-Eisenhauer, A. (2008). "Morphology of the sella turcica in Axenfeld-Rieger syndrome with PITX2 mutation". Journal of Oral Pathology & Medicine. 37 (8): 504–10. doi:10.1111/j.1600-0714.2008.00650.x. PMID 18331556.
- 1 2 3 Axenfeld, T (1920). "Embryotoxon cornea posterius". Berichte der Deutschen Ophthalmologischen Gesellschaft. 42: 301–2.
- ↑ John F., Salmon (2020). "Glaucoma". Kanski's clinical ophthalmology : a systematic approach (9th ed.). Edinburgh: Elsevier. ISBN 978-0-7020-7713-5. OCLC 1131846767.
- 1 2 3 Lowry, R. Brian; Gould, Douglas B.; Walter, Michael A.; Savage, Paul R. (2007). "Absence of PITX2, BARX1, and FOXC1 mutations in De Hauwere syndrome (Axenfeld–Rieger anomaly, hydrocephaly, hearing loss): A 25-year follow up". American Journal of Medical Genetics Part A. 143A (11): 1227–30. doi:10.1002/ajmg.a.31732. PMID 17486624.
- ↑ Reis, LM; Semina, EV (September 2011). "Genetics of anterior segment dysgenesis disorders". Current Opinion in Ophthalmology. 22 (5): 314–24. doi:10.1097/ICU.0b013e328349412b. PMC 3558283. PMID 21730847.
- 1 2 "Axenfeld-Rieger syndrome". United States Department of Health and Human Services. November 8, 2016. Archived from the original on August 9, 2020. Retrieved September 19, 2021.
- 1 2 Phillips, Jeffrey C.; del Bono, Elizabeth A.; Haines, Jonathan L.; Pralea, Anca Madalina; Cohen, John S.; Greff, Linda J.; Wiggs, Janey L. (1996). "A second locus for Rieger syndrome maps to chromosome 13q14". American Journal of Human Genetics. 59 (3): 613–9. PMC 1914897. PMID 8751862.
- ↑ "How are genetic conditions diagnosed?". United States Department of Health and Human Services. November 8, 2016. Archived from the original on September 19, 2020. Retrieved September 19, 2021.
- ↑ synd/1284 at Who Named It?
Further reading
- Amendt, Brad A., ed. (2005). The Molecular Mechanisms of Axenfeld-Rieger Syndrome. Medical Intelligence Unit. Springer. doi:10.1007/0-387-28672-1. ISBN 978-0-387-28672-3.
- Agarwal, Sunita; Agarwal, Athiya; Apple, David J., eds. (2002). "Axenfeld-Rieger Syndrome". Textbook of Ophthalmology. Jaypee Brothers. pp. 1049–51. ISBN 978-81-7179-884-1.
{{cite book}}: CS1 maint: url-status (link) - Shields, M.Bruce; Buckley, Edward; Klintworth, Gordon K.; Thresher, Randy (1985). "Axenfeld-Rieger syndrome. A spectrum of developmental disorders". Survey of Ophthalmology. 29 (6): 387–409. doi:10.1016/0039-6257(85)90205-X. PMID 3892740.
- Alward, Wallace L.M. (2000). "Axenfeld-Rieger syndrome in the age of molecular genetics". American Journal of Ophthalmology. 130 (1): 107–15. doi:10.1016/S0002-9394(00)00525-0. PMID 11004268.
External links
| Classification |
|---|
- Axenfeld Rieger syndrome at NIH's Office of Rare Diseases
- Axenfeld Rieger anomaly with cardiac defects and sensorineural hearing loss at NIH's Office of Rare Diseases