Hemophagocytic lymphohistiocytosis
| Hemophagocytic lymphohistiocytosis | |
|---|---|
| Other names: Haemophagocytic lymphohistiocytosis (British spelling), hemophagocytic or haemophagocytic syndrome,[1] familial hemophagocytic reticulosis[2] | |
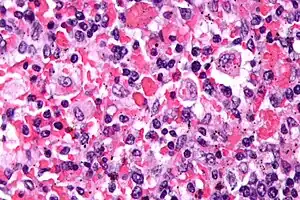 | |
| Micrograph showing red blood cells within macrophages. H&E stain. | |
| Specialty | Hematology |
| Symptoms | Fever, bleeding problems, liver problems, large spleen, low blood cells[3] |
| Complications | Acute respiratory distress syndrome (ARDS), myocarditis, liver failure, encephalitis[4] |
| Usual onset | Early childhood, 50s[4] |
| Types | Primary (genetic), secondary[4] |
| Causes | Genetic mutations, cancer, infection, autoimmune problems[3] |
| Diagnostic method | Based on symptoms, blood tests, medical imaging, tissue biopsy[4] |
| Differential diagnosis | Sepsis, Kawasaki disease, toxic shock syndrome[4] |
| Treatment | Etoposide, immunosuppressants, stem cell transplantation[3] |
| Frequency | Rare[5] |
| Deaths | 22% 5-year survival (children)[4] |
Hemophagocytic lymphohistiocytosis (HLH) is a blood disorder in which the body produces high levels of cytokines resulting in organ damage.[3] Symptoms include fever, bleeding problems, liver problems, a large spleen, and low blood cells.[3] Complications can include acute respiratory distress syndrome (ARDS), myocarditis, liver failure, and encephalitis.[4]
It most commonly occurs due to inherited genetic mutations (25%) that alter regulation of the immune system or secondary to cancer, infection, or autoimmune problems.[3][5] The underlying mechanism involves activation of cytotoxic T lymphocytes, natural killer (NK) cells, and macrophages.[3] Diagnosis is based on symptoms, blood tests, medical imaging, and possibly tissue biopsy.[4] It is a type of cytokine storm.[6]
Treatment is with etoposide, immunosuppressants, such as corticosteroids, and stem cell transplantation.[3][7] Blood transfusion or platelet transfusion may be required.[5] In children, despite treatment, the 5-year survival rate is about 22%, while the risk of death in adults is about 40%.[4]
Hemophagocytic lymphohistiocytosis is rare, with the genetic form affecting about 1.5 per million people a year.[4] Other forms occur in about one in 2,000 people in the intensive care unit.[4] Onset is typically either in early childhood or around the age of 50.[4] Among children both sexes are affected with similar frequency.[4] The condition was first described in 1952 by Farquhar and Claireaux.[2]
Signs and symptoms
The onset of HLH occurs before the age of one year in approximately 70 percent of cases. Familial HLH should be suspected if siblings are diagnosed with HLH or if symptoms recur when therapy has been stopped. Familial HLH is an autosomal recessive disease, hence each sibling of a child with familial HLH has a twenty-five–percent chance of developing the disease, a fifty-percent chance of carrying the defective gene (which is very rarely associated with any risk of disease), and a twenty-five–percent chance of not being affected and not carrying the gene defect.[8]
Symptoms commonly include fever, enlargement of the liver and spleen, enlarged lymph nodes, yellow discoloration of the skin and eyes, and a rash.[9] Laboratory findings may include elevated triglyceride levels, low fibrinogen levels, transaminitis, and elevated ferritin levels (among others).[9]
Blood changes may include cytopenia and hyperferritinemia.[10] In the earlier stages of HLH are frequently hospitalized at internal medicine wards.[11]
Causes
Primary HLH is caused by loss of function, (i.e. inactivating) mutations in genes that code for proteins cytotoxic T cells and NK cells use to kill targeted cells, such as those infected with pathogens like the Epstein-Barr virus (EBV) or the Dengue virus.[12] These mutations include those in the following genes: UNC13D, STX11, RAB27A, STXBP2, LYST, PRF1 1, SH2D1A, BIRC4, ITK, CD27, and MAGT1.[13]
Secondary HLH (sHLH) is associated with, and thought to be promoted, by malignant and non-malignant diseases that likewise weaken the ability of the immune system to attack EBV-infected cells. Malignant disorders associated with secondary HLH include T-cell lymphoma, B-cell lymphoma, acute lymphocytic leukemia, acute myeloid leukemia, and myelodysplastic syndrome. Non-malignant disorders associated with secondary HLH include: autoimmune disorders such as juvenile idiopathic arthritis, juvenile Kawasaki disease, systemic lupus erythematosus, the juvenile onset and adult onset forms of Still's disease, and rheumatoid arthritis;[13] immunodeficiency disorders such as severe combined immunodeficiency, DiGeorge syndrome, Wiskott–Aldrich syndrome, ataxia–telangiectasia, and dyskeratosis congenita);[14] and infections caused by EBV, cytomegalovirus, HIV/AIDS, bacteria, protozoa, fungi and possibly SARS-CoV-2.[15] Secondary HLH may also result from iatrogenic causes such as bone marrow or other organ transplantations; chemotherapy; or therapy with immunosuppressing agents;[16]
About 33% of all HLH cases, ~75% of Asian HLH cases, and nearly 100% of HLH cases caused by mutations in SH2D1A (see X-linked lymphoproliferative disease type 1) are associated with, and thought triggered or promoted by, EBV infection. These cases of HLH are classified as belonging to the class of Epstein–Barr virus-associated lymphoproliferative diseases and termed EBV+ HLH.[17]
Genetics
Five genetic subtypes (FHL1, FHL2, FHL3, FHL4, and FHL5) are described, with an estimated overall prevalence of one in 50,000 and equal gender distribution. Molecular genetic testing for four of the causative genes, PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5), is available on a clinical basis. Symptoms of FHL are usually evident within the first few months of life and may even develop in utero. However, symptomatic presentation throughout childhood and even into young adulthood has been observed in some cases.
The five subtypes of FHL[18] are each associated with a specific gene:
- FHL1: HPLH1
- FHL2: PRF1 (Perforin)
- FHL3: UNC13D (Munc13-4)
- FHL4: STX11 (Syntaxin 11)
- FHL5: STXBP2 (Syntaxin binding protein 2)/UNC18-2
Nearly half of the cases of type 2 familial hemophagocytic lymphohistiocytosis are due to bi-allelic PRF1 mutations.[19]
Pathophysiology
The underlying causes, either inherited or acquired, lead to an unchecked immune response when exposed to triggers. Impaired NK-cell cytotoxicity is the hallmark of HLH. All genetic defects for familial HLH are related to granule-dependent cytotoxicity. This inability to remove infected and antigen-presenting cells and terminate the immune response leads to uncontrolled proliferation and activation of the immune system with release of excessive cytokines. These cells then infiltrate organs, releasing more cytokines, which gives the clinical picture. The fever is caused by IL-1, IL-6 and TNF-alpha; the cytopenia is due to the suppressive effect on hematopoiesis by TNF-alpha and TNF-gamma. TNF-alpha and TNF-gamma may also lead to inhibition of lipoprotein lipase or stimulate triglyceride synthesis. Activated macrophages secrete ferritin and plasminogen activator leading to hyperfibrinolysis.[20]
Diagnosis
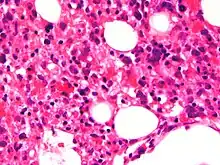
The blood count typically shows decreased numbers of blood cells—including a decreased number of circulating red blood cells, white blood cells, and platelets. The bone marrow may show hemophagocytosis. The liver function tests are usually elevated. A low level of the protein albumin in the blood is common.
The serum C reactive protein, erythrocyte sedimentation rate, and ferritin level are markedly elevated. In children, a ferritin above 10000 is very sensitive and specific for the diagnosis of HLH,[21] however, the diagnostic utility for ferritin is less for adult HLH patients.[22]
The serum fibrinogen level is usually low and the D-dimer level is elevated.
The sphingomyelinase is elevated.[23]
Bone marrow biopsy shows histiocytosis.[24]
Classification
Primary HLH, also known as familial haemophagocytic lymphohistiocytosis (FHL) or familial erythrophagocytic lymphohistiocytosis, is a heterogeneous autosomal recessive disorder found to be more prevalent with parental consanguinity.
Secondary haemophagocytic lymphohistiocytosis (acquired haemophagocytic lymphohistiocytosis) occurs after strong immunologic activation, such as that which can occur with systemic infection, immunodeficiency, or underlying malignancy.[25]
Both forms are characterized by the overwhelming activation of normal T lymphocytes and macrophages, invariably leading to clinical and haematologic alterations and death in the absence of treatment.
A subtype of primary HLH where the inflammation is limited to the central nervous system has been described.[26]
Diagnostic criteria
The current (2008) diagnostic criteria for HLH are[27]
1. A molecular diagnosis consistent with HLH. These include the identification of pathologic mutations of PRF1, UNC13D, or STX11.
OR
2. Fulfillment of five out of the eight criteria below:
- Fever (defined as a temperature >100.3 °F, >38 °C)
- Enlargement of the spleen
- Decreased blood cell counts affecting at least two of three lineages in the peripheral blood:
- Haemoglobin <9 g/100 ml (in infants <4 weeks: haemoglobin <10 g/100 ml) (anemia)
- Platelets <100×109/L (thrombocytopenia)
- Neutrophils <1×109/L (neutropenia)
- High blood levels of triglycerides (fasting, greater than or equal to 265 mg/100 ml) and/or decreased amounts of fibrinogen in the blood (≤ 150 mg/100 ml)
- Ferritin ≥ 500 ng/ml
- Haemophagocytosis in the bone marrow, spleen or lymph nodes
- Low or absent natural killer cell activity
- Soluble CD25 (soluble IL-2 receptor) >2400 U/ml (or per local reference laboratory)
In addition, in the case of familial HLH, no evidence of malignancy should be apparent.
Not all five out of eight criteria are required for diagnosis of HLH in adults, and a high index of suspicion is required for diagnosis as delay results in increased mortality. The diagnostic criteria were developed in pediatric populations and have not been validated for adult HLH patients.[28] Attempts to improve diagnosis of HLH have included use of the HScore Archived 2015-12-22 at the Wayback Machine, which can be used to estimate an individual's risk of HLH.[29] In adults, soluble IL-2 receptor has been found to be a very sensitive marker for HLH, demonstrating 100% sensitivity for ruling out HLH below a cutoff of 2400 U/mL and optimal cutoff for ruling in at 2515 U/mL (sensitivity, 100%; specificity, 72.5%), with 93% specificity at >10 000 U/mL.[30]
Differential diagnosis
The differential diagnosis of HLH includes secondary HLH and macrophage-activation syndrome or other primary immunodeficiencies that present with hemophagocytic lymphohistiocytosis, such as X-linked lymphoproliferative disease.
Other conditions that may be confused with this condition include autoimmune lymphoproliferative syndrome.[31] As a syndrome of intense inflammation it needs to be differentiated from sepsis, what may be extremely challenging.[32]
The diagnosis of acquired, or secondary, HLH is usually made in association with infection by viruses, bacteria, fungi, or parasites or in association with lymphoma, autoimmune disease, or metabolic disease. Acquired HLH may have decreased, normal, or increased NK cell activity.
Griscelli syndrome
A major differential diagnosis of HLH is Griscelli syndrome (type 2). This is a rare autosomal recessive disorder characterized by partial albinism, hepatosplenomegaly, pancytopenia, hepatitis, immunologic abnormalities, and lymphohistiocytosis. Most cases have been diagnosed between 4 months and 7 years of age, with a mean age of about 17 months.
Three types of Griscelli syndrome are recognised: type 1 has neurologic symptoms and mutations in MYO5A. Prognosis depends on the severity of neurologic manifestations. Type 2 has mutations in RAB27A and haemophagocytic syndrome, with abnormal T-cell and macrophage activation. This type has a grave prognosis if untreated. Type 3 has mutations in melanophilin and is characterized by partial albinism. This type does not pose a threat to those so affected.
Treatment
In secondary cases, treatment of the cause, where possible, is indicated. Additionally, treatment for HLH itself is usually required.
While optimal treatment of HLH is still being debated, current treatment regimes usually involve high dose corticosteroids, etoposide and cyclosporin. Intravenous immunoglobulin is also used. Methotrexate and vincristine have also been used. Other medications include cytokine targeted therapy.
On 20 November 2018, the FDA approved the anti-IFN-gamma monoclonal antibody emapalumab (proprietary name Gamifant) for the treatment of pediatric and adult primary HLH.[33]
In October 2021 NHS England published Clinical Commissioning Policy: Anakinra for Haemophagocytic Lymphohistiocytosis (HLH) for adults and children in all ages, allowing Anakinra (a modified recombinant interleukin 1 receptor antagonist) to be used in the treatment of HLH.[34]
Prognosis
The prognosis is guarded with an overall mortality of 50%. Poor prognostic factors included HLH associated with malignancy, with half the patients dying by 1.4 months compared to 22.8 months for non-tumour associated HLH patients.[35]
Secondary HLH in some individuals may be self-limited because patients are able to fully recover after having received only supportive medical treatment (i.e., IV immunoglobulin only). However, long-term remission without the use of cytotoxic and immune-suppressive therapies is unlikely in the majority of adults with HLH and in those with involvement of the central nervous system (brain and/or spinal cord).[18]
History
The first case report of HLH was published in 1952.[36]
Research
A systematic review recently reported the pooled proportion are fever 97.2%, hepatomegaly 70.2%, splenomegaly 78.4%, thrombocytopenia 90.1%, anemia 76.0%, and serum ferritin ≥500 μg/L 97.1%. The case fatality rate is 14.6% among dengue hemophagocytic lymphohistiocytosis patients.[37]
See also
References
- ↑ Fisman, David N. (2000). "Hemophagocytic syndromes and infection". Emerging Infect. Dis. 6 (6): 601–8. doi:10.3201/eid0606.000608. PMC 2640913. PMID 11076718.
- 1 2 Rosado, Flavia G. N.; Kim, Annette S. (June 2013). "Hemophagocytic Lymphohistiocytosis: An Update on Diagnosis and Pathogenesis". American Journal of Clinical Pathology. 139 (6): 713–727. doi:10.1309/AJCP4ZDKJ4ICOUAT.
- 1 2 3 4 5 6 7 8 Al-Samkari, H; Berliner, N (24 January 2018). "Hemophagocytic Lymphohistiocytosis". Annual review of pathology. 13: 27–49. doi:10.1146/annurev-pathol-020117-043625. PMID 28934563.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Konkol, S; Rai, M (January 2022). "Lymphohistiocytosis". StatPearls. PMID 32491708.
- 1 2 3 "Hemophagocytic Lymphohistiocytosis". NORD (National Organization for Rare Disorders). Archived from the original on 10 May 2022. Retrieved 9 December 2022.
- ↑ Kliegman, Robert M.; III, Joseph W. St Geme (1 April 2019). Nelson Textbook of Pediatrics E-Book. Elsevier Health Sciences. p. 2709. ISBN 978-0-323-56888-3. Archived from the original on 9 December 2022. Retrieved 9 December 2022.
- ↑ Skinner, J; Yankey, B; Shelton, BK (2019). "Hemophagocytic Lymphohistiocytosis". AACN advanced critical care. 30 (2): 151–164. doi:10.4037/aacnacc2019463. PMID 31151946.
- ↑ "Familial hemophagocytic lymphohistiocytosis: MedlinePlus Genetics". medlineplus.gov. Archived from the original on 2021-01-25. Retrieved 2021-01-24.
- 1 2 Esteban, Ysabella M.; de Jong, Jill L. O.; Tesher, Melissa S. (1 August 2017). "An Overview of Hemophagocytic Lymphohistiocytosis". Pediatric Annals. 46 (8): e309–e313. doi:10.3928/19382359-20170717-01. PMID 28806468.
- ↑ Machowicz, Rafal; Janka, Gritta; Wiktor-Jedrzejczak, Wieslaw (2016-01-01). "Your critical care patient may have HLH (hemophagocytic lymphohistiocytosis)". Critical Care. 20 (1): 215. doi:10.1186/s13054-016-1369-3. ISSN 1364-8535. PMC 4937543. PMID 27389585.
- ↑ Machowicz, Rafal; Basak, Grzegorz (2020-03-05). "How can an internal medicine specialist save a patient with hemophagocytic lymphohistiocytosis (HLH)?". Polish Archives of Internal Medicine. 130 (5): 431–437. doi:10.20452/pamw.15226. PMID 32134401.
- ↑ Giang HT, Banno K, Minh LH, Trinh LT, Loc LT, Eltobgy A, Tai LL, Khan A, Tuan NH, Reda Y, Samsom M. Dengue hemophagocytic syndrome: A systematic review and meta‐analysis on epidemiology, clinical signs, outcomes, and risk factors. Reviews in medical virology. 2018 Nov;28(6):e2005. https://onlinelibrary.wiley.com/doi/abs/10.1002/rmv.2005 Archived 2020-06-07 at the Wayback Machine https://doi.org/10.1002/rmv.2005 Archived 2021-10-18 at the Wayback Machine
- 1 2 Wysocki CA (December 2017). "Comparing hemophagocytic lymphohistiocytosis in pediatric and adult patients". Current Opinion in Allergy and Clinical Immunology. 17 (6): 405–413. doi:10.1097/ACI.0000000000000405. PMID 28957822. S2CID 11439142.
- ↑ Bode SF, Ammann S, Al-Herz W, Bataneant M, Dvorak CC, Gehring S, Gennery A, Gilmour KC, Gonzalez-Granado LI, Groß-Wieltsch U, Ifversen M, Lingman-Framme J, Matthes-Martin S, Mesters R, Meyts I, van Montfrans JM, Pachlopnik Schmid J, Pai SY, Soler-Palacin P, Schuermann U, Schuster V, Seidel MG, Speckmann C, Stepensky P, Sykora KW, Tesi B, Vraetz T, Waruiru C, Bryceson YT, Moshous D, Lehmberg K, Jordan MB, Ehl S (July 2015). "The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis". Haematologica. 100 (7): 978–88. doi:10.3324/haematol.2014.121608. PMC 4486233. PMID 26022711.
- ↑ Mehta, Puja; McAuley, Daniel F.; Brown, Michael; Sanchez, Emilie; Tattersall, Rachel S.; Manson, Jessica J. (2020-03-28). "COVID-19: consider cytokine storm syndromes and immunosuppression". The Lancet. 395 (10229): 1033–1034. doi:10.1016/S0140-6736(20)30628-0. ISSN 0140-6736. PMC 7270045. PMID 32192578.
- ↑ Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, Blechacz B, Wang S, Minkov M, Jordan MB, La Rosée P, Kantarjian HM (September 2017). "A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults". Cancer. 123 (17): 3229–3240. doi:10.1002/cncr.30826. PMC 5568927. PMID 28621800.
- ↑ Marsh RA (2017). "Epstein–Barr Virus and Hemophagocytic Lymphohistiocytosis". Frontiers in Immunology. 8: 1902. doi:10.3389/fimmu.2017.01902. PMC 5766650. PMID 29358936.
- 1 2 Zhang, Kejian; Filopovich, Alexandra H.; Johnson, Judith; Marsh, Rebecca A.; Villanueva, Joyce (January 17, 2013). "Hemophagocytic Lymphohistiocytosis, Familial". GeneReviews. PMID 20301617. NBK1444. Archived from the original on October 18, 2021. Retrieved October 14, 2021.
- ↑ Trapani JA, Thia KY, Andrews M, et al. (April 2013). "Human perforin mutations and susceptibility to multiple primary cancers". Oncoimmunology. 2 (4): e24185. doi:10.4161/onci.24185. PMC 3654607. PMID 23734337.
- ↑ Usmani, G. Naheed; Woda, Bruce A.; Newburger, Peter E. (2013). "Advances in understanding the pathogenesis of HLH". British Journal of Haematology. 161 (5): 609–622. doi:10.1111/bjh.12293. ISSN 1365-2141. PMID 23577835.
- ↑ Allen, Carl (June 2008). "Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis". Pediatric Blood & Cancer. 50 (6): 1227–35. doi:10.1002/pbc.21423. PMID 18085676.
- ↑ Schram, Alison (March 5, 2015). "Marked hyperferritinemia does not predict for HLH in the adult population". Blood. 125 (10): 1548–52. doi:10.1182/blood-2014-10-602607. PMID 25573993.
- ↑ Jenkins RW, Clarke CJ, Lucas JT, et al. (November 2013). "Evaluation of the role of secretory sphingomyelinase and bioactive sphingolipids as biomarkers in hemophagocytic lymphohistiocytosis". Am. J. Hematol. 88 (11): E265–72. doi:10.1002/ajh.23535. PMC 4348111. PMID 23828274.
- ↑ Lymphohistiocytosis,+Hemophagocytic at the US National Library of Medicine Medical Subject Headings (MeSH)
- ↑ Ponnatt, Tanya Sajan; Lilley, Cullen M.; Mirza, Kamran M. (2021-08-04). "Hemophagocytic Lymphohistiocytosis". Archives of Pathology & Laboratory Medicine. doi:10.5858/arpa.2020-0802-RA. ISSN 1543-2165. PMID 34347856. Archived from the original on 2021-08-05. Retrieved 2021-10-14.
- ↑ Benson LA, Li H, Henderson LA, Solomon IH, Soldatos A, Murphy J, Bielekova B, Kennedy AL, Rivkin MJ, Davies KJ, Hsu AP, Holland SM, Gahl WA, Sundel RP, Lehmann LE, Lee MA, Alexandrescu S, Degar BA, Duncan CN, Gorman MP (2019) Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm 6(3):e560
- ↑ Jordan MB, Filipovich AH (October 2008). "Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: a journey of a thousand miles begins with a single (big) step". Bone Marrow Transplant. 42 (7): 433–7. doi:10.1038/bmt.2008.232. PMID 18679369.
- ↑ Schram, Alison (May 7, 2015). "How I treat hemophagocytic lymphohistiocytosis in the adult patient". Blood. 125 (19): 2908–14. doi:10.1182/blood-2015-01-551622. PMID 25758828.
- ↑ Fardet, Laurence (September 9, 2014). "Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome". Arthritis & Rheumatology. 66 (9): 2613–20. doi:10.1002/art.38690. PMID 24782338.
- ↑ Hayden, Anna (December 2017). "Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH". Blood Advances. 1 (26): 2529–2534. doi:10.1182/bloodadvances.2017012310. PMC 5728644. PMID 29296904.
- ↑ Rudman Spergel A, Walkovich K, Price S, et al. (November 2013). "Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis". Pediatrics. 132 (5): e1440–4. doi:10.1542/peds.2012-2748. PMC 3813387. PMID 24101757.
- ↑ Machowicz R, Janka G, Wiktor-Jedrzejczak W (March 2017). "Similar but not the same: Differential diagnosis of HLH and sepsis". Critical Reviews in Oncology/Hematology. 114: 1–12. doi:10.1016/j.critrevonc.2017.03.023. PMID 28477737.
- ↑ "Press Announcements - FDA approves first treatment specifically for patients with rare and life-threatening type of immune disease". 2019-03-06. Archived from the original on 2019-04-23. Retrieved 2021-10-14.
- ↑ NHS England (October 2021). Clinical Commissioning Policy: Anakinra for Haemophagocytic Lymphohistiocytosis (HLH) for adults and children in all ages (PDF). Archived from the original (PDF) on 13 October 2021. Retrieved 14 October 2021.
- ↑ Parikh, Sameer (April 2014). "Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis". Mayo Clinic Proceedings. 89 (4): 484–92. doi:10.1016/j.mayocp.2013.12.012. PMID 24581757. Archived from the original on October 18, 2021. Retrieved December 14, 2015.
- ↑ Farquhar, James W.; Claireaux, Albert E. (December 1952). "Familial Haemophagocytic Reticulosis". Archives of Disease in Childhood. 27 (136): 519–525. doi:10.1136/adc.27.136.519. PMC 1988563. PMID 13008468.
- ↑ Giang, Hoang Thi Nam; Banno, Keita; Minh, Le Huu Nhat; Trinh, Lam Tuyet; Loc, Le Thai; Eltobgy, Asmaa; Tai, Luu Lam Thang; Khan, Adnan; Tuan, Nguyen Hoang (2018-08-15). "Dengue hemophagocytic syndrome: A systematic review and meta-analysis on epidemiology, clinical signs, outcomes, and risk factors". Reviews in Medical Virology. 28 (6): e2005. doi:10.1002/rmv.2005. ISSN 1052-9276. PMID 30109914. S2CID 52002485.
External links
| Classification | |
|---|---|
| External resources |
|