Hartnup disease
| Hartnup disease | |
|---|---|
| Other names: Aminoaciduria, Hartnup type | |
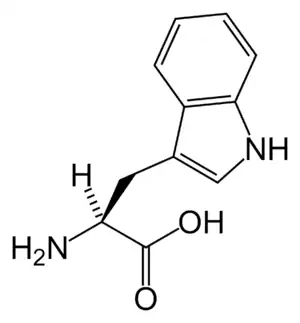 | |
| Tryptophan | |
Hartnup disease (also known as "pellagra-like dermatosis"[1] and "Hartnup disorder"[2]) is an autosomal recessive[3] metabolic disorder affecting the absorption of nonpolar amino acids (particularly tryptophan that can be, in turn, converted into serotonin, melatonin, and niacin). Niacin is a precursor to nicotinamide, a necessary component of NAD+.[4]: 541
The causative gene, SLC6A19, is located on chromosome 5.[5] It is named after the British family, Hartnup, who suffered from this disease.
Signs and symptoms
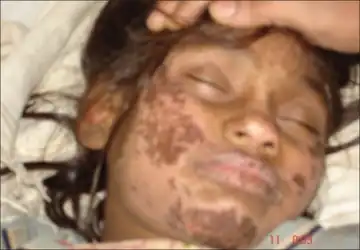
Hartnup disease manifests during infancy with variable clinical presentation: failure to thrive, photosensitivity, intermittent ataxia, nystagmus, and tremor.
Nicotinamide is necessary for neutral amino acid transporter production in the proximal renal tubules found in the kidney, and intestinal mucosal cells found in the small intestine. Therefore, a symptom stemming from this disorder results in increased amounts of amino acids in the urine. Pellagra, a similar condition, is also caused by low nicotinamide; this disorder results in dermatitis, diarrhea, and dementia.
Hartnup disease is a disorder of amino acid transport in the intestine and kidneys; otherwise, the intestine and kidneys function normally, and the effects of the disease occur mainly in the brain and skin. Symptoms may begin in infancy or early childhood, but sometimes they begin as late as early adulthood. Symptoms may be triggered by sunlight, fever, drugs, or emotional or physical stress. A period of poor nutrition nearly always precedes an attack. The attacks usually become progressively less frequent with age. Most symptoms occur sporadically and are caused by a deficiency of niacinamide. A rash develops on parts of the body exposed to the sun. Mental retardation, short stature, headaches, unsteady gait, and collapsing or fainting are common. Psychiatric problems (such as anxiety, rapid mood changes, delusions, and hallucinations) may also result.[6]
Causes
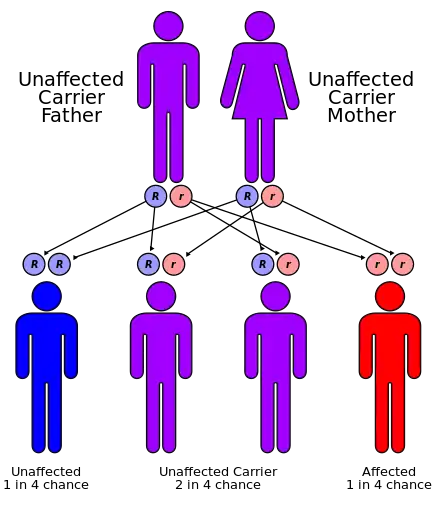
Hartnup disease is inherited as an autosomal recessive trait. Heterozygotes are normal. Consanguinity is common. The failure of amino-acid transport was reported in 1960 from the increased presence of indoles (bacterial metabolites of tryptophan) and tryptophan in the urine of patients as part of a generalized aminoaciduria of the disease. The excessive loss of tryptophan from malabsorption was the cause of the pellagra like symptoms. From studies on ingestion of tryptophan it seemed that there was a generalized problem with amino-acid transport.[7] In 2004, a causative gene, SLC6A19, was located on band 5p15.33. SLC6A19 is a sodium-dependent and chloride-independent neutral amino acid transporter, expressed predominately in the kidneys and intestine.[8]
Diagnosis
The defective gene controls the absorption of certain amino acids from the intestine and the reabsorption of those amino acids in the kidneys. Consequently, a person with Hartnup disease cannot absorb amino acids properly from the intestine and cannot reabsorb them properly from tubules in the kidneys. Excessive amounts of amino acids, such as tryptophan, are excreted in the urine. The body is thus left with inadequate amounts of amino acids, which are the building blocks of proteins. With too little tryptophan in the blood, the body is unable to make a sufficient amount of the B-complex vitamin niacinamide, particularly under stress when more vitamins are needed.[6]
In Hartnup disease, urinary excretion of proline, hydroxyproline, and arginine remains unchanged, differentiating it from other causes of generalized aminoaciduria, such as Fanconi syndrome. With urine chromatography, increased levels of neutral amino acids (e.g., glutamine, valine, phenylalanine, leucine, asparagine, citrulline, isoleucine, threonine, alanine, serine, histidine, tyrosine, tryptophan) and indican are found in the urine. Increased urinary Indican can be tested by Obermeyer test.
Treatment
A high-protein diet can overcome the deficient transport of neutral amino acids in most patients. Poor nutrition leads to more frequent and more severe attacks of the disease, which is otherwise asymptomatic. All patients who are symptomatic are advised to use physical and chemical protection from sunlight: avoid excessive exposure to sunlight, wear protective clothing, and use chemical sunscreens with a SPF of 15 or greater. Patients also should avoid other aggravating factors, such as photosensitizing drugs, as much as possible. In patients with niacin deficiency and symptomatic disease, daily supplementation with nicotinic acid or nicotinamide reduces both the number and severity of attacks. Neurologic and psychiatric treatment is needed in patients with severe central nervous system involvement.[8]
See also
References
- ↑ Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ↑ Online Mendelian Inheritance in Man (OMIM): 234500
- ↑ Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, Arcos-Burgos M, Dave MH, Wagner CA, Camargo SR, Inoue S, Matsuura N, Helip-Wooley A, Bockenhauer D, Warth R, Bernardini I, Visser G, Eggermann T, Lee P, Chairoungdua A, Jutabha P, Babu E, Nilwarangkoon S, Anzai N, Kanai Y, Verrey F, Gahl WA, Koizumi A (September 2004). "Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder". Nature Genetics. 36 (9): 999–1002. doi:10.1038/ng1405. PMID 15286787. S2CID 155361.
- ↑ James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- ↑ Seow HF, Brer S, Brer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE (September 2004). "Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19". Nature Genetics. 36 (9): 1003–7. doi:10.1038/ng1406. PMID 15286788.
- 1 2 LaRosa, CJ (January 2020). "Hartnup Disease". Archived from the original on 8 July 2020. Retrieved 6 July 2020.
- ↑ Milne, M.D., Crawford, M.A., Girao, C.B. and Loughridge, L. (1961) The metabolic disorder of the Hartnup disease. Q. J. Med. 29: 407-421
- 1 2 Sekulovic, LJ (February 2017). "Hartnup Disease". Archived from the original on 23 November 2008. Retrieved 6 July 2020.
External links
| Classification | |
|---|---|
| External resources |
|