Neurofibromatosis type II
| Neurofibromatosis type II | |
|---|---|
| Other names: multiple inherited schwannomas, meningiomas, and ependymomas (MISME syndrome)) | |
 | |
| Micrograph of a schwannoma, a tumor seen in neurofibromatosis type II. HPS stain. | |
Neurofibromatosis type II (also known as MISME syndrome – multiple inherited schwannomas, meningiomas, and ependymomas) is a genetic condition that may be inherited or may arise spontaneously. The main manifestation of the condition is the development of bilateral benign brain tumors in the nerve sheath of the cranial nerve VIII, which is the "auditory-vestibular nerve" that transmits sensory information from the inner ear to the brain. Besides, other benign brain and spinal tumors occur. Symptoms depend on the presence, localisation and growth of the tumor(s). Many people with this condition also experience vision problems. Neurofibromatosis type II (NF2 or NF II) is caused by mutations of the "Merlin" gene,[1] which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions. Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation.
Classification
NF2 is an inheritable disorder with an autosomal dominant mode of transmission.[2]
There are two forms of the NF2:[3]
- The Wishart-Phenotype is characterized by multiple cerebral and spinal lesions in people younger than 20 years and with rapid progression of the tumours.
- People with NF2 who develop single central tumours with slow progression after the age of 20 are thought to have the Feiling-Gardner-Phenotype.
Symptoms and signs

The clinical presentation of Neurofibromatosis type II is consistent with the following:[4]
- Neuroma
- Wide-set eyes
- Double-Jointed
- Posterior subcapsular cataract
- Short stature
- Blindness
- Diplopia
- Dysarthria
- Epicanthus
Cause

NF2 is caused by inactivating mutations in the NF2 gene located at 22q12.2 of chromosome 22, type of mutations vary and include protein-truncating alterations (frameshift deletions/insertions and nonsense mutations), splice-site mutations, missense mutations and others. Deletions, too, in the NH2-terminal domain of merlin proteins have been associated with early tumor onset and poor prognosis in people with NF2.[5] Protein truncating mutations correlate with more severe phenotype.[6] There is a broad clinical spectrum known, but all people with the condition who have been checked have been found to have some mutation of the same gene on chromosome 22. Through statistics, it is suspected that one-half of cases are inherited, and one-half are the result of new, de novo mutations.
Pathogenesis
NF2 is caused by a defect in the gene that normally gives rise to a product called Merlin or Schwannomin, located on chromosome 22 band q11-13.1. Merlin was first discovered as a structural protein functioning as an actin cytoskeleton regulator. Later merlin's tumour suppressant role was described. Merlin regulates multiple proliferative signalling cascades such as receptor tyrosine kinase signalling, p21-activated kinase signalling, Ras signalling, MEK-ERK cascade, MST-YAP cascade.[7] In a normal cell, the concentrations of active (dephosphorylated) merlin are controlled by processes such as cell adhesion (which would indicate the need to restrain cell division). It has been shown that Merlin inhibits Rac1 which is crucial for cell motility and tumour invasion.[8] Also, merlin's interaction with cyclin D was described.[9] It is known that Merlin's deficiency can result in unmediated progression through the cell cycle due to the lack of contact-mediated tumour suppression, mainly because of the cell:cell junction disruption, sufficient to result in the tumors characteristic of Neurofibromatosis type II. Recent studies showed that besides its cytoskeletal and cytoplasmic functions Merlin also translocates to the nucleus and suppresses proliferation by inhibiting E3 ubiquitin ligase CRL4(DCAF1).[10] Finally, most recent studies indicated that Merlin also plays important role in energy metabolism regulation.[11][12] Mutations of NF2 is presumed to result in either a failure to synthesize Merlin or the production of a defective peptide that lacks the normal tumor-suppressive effect. The Schwannomin-peptide consists of 595 amino acids. Comparison of Schwannomin with other proteins shows similarities to proteins that connect the cytoskeleton to the cell membrane. Mutations in the Schwannomin-gene are thought to alter the movement and shape of affected cells with loss of contact inhibition. Ependymomas are tumors arising from the ependyma, an epithelium-like tissue of the central nervous system.[13] In people with NF2 and ependymomas, the tumor suppressant function of Merlin may be compromised. Loss of function mutations occurring in chromosome 22q, where Merlin proteins are coded, can promote tumorigenesis, or the creation of new tumorous cells.[5] Deletions, too, in the NH2-terminal domain of merlin proteins have been associated with early tumor onset and poor prognosis in affected people.[5]
Pathology
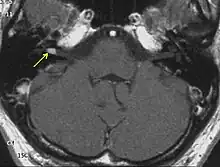
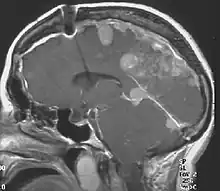
The so-called acoustic neuroma of NF2 is in fact a schwannoma of the nervus vestibularis, or vestibular schwannoma. The misnomer of acoustic neuroma is still often used. The vestibular schwannomas grow slowly at the inner entrance of the internal auditory meatus (meatus acousticus internus). They derive from the nerve sheaths of the upper part of the nervus vestibularis in the region between the central and peripheral myelin (Obersteiner-Redlich-Zone) within the area of the porus acousticus, 1 cm from the brainstem.
Genotype-phenotype-correlation
Many people with NF2 were included in studies that were designed to compare disease type and progression with exact determination of the associated mutation. The goal of such comparisons of genotype and phenotype is to determine whether specific mutations cause respective combinations of symptoms. This would be extremely valuable for the prediction of disease progression and the planning of therapy starting at a young age. The results of such studies are the following:
- In most cases the mutation in the NF2 gene causes shortened peptides.
- There are no mutational hot spots.
- People with frameshift mutations or nonsense mutations have poor prognosis.
- People with missense mutations have a better prognosis.
- In cases with mutations in the splice-acceptor-region, there is no good correlation to determine.
- Point mutations may have only minor effects.
- Cases are published in which exactly the same mutation is associated with clearly different outcomes.
These results suggest that other factors (environment, other mutations) will probably determine the clinical outcome.
Diagnosis

Prenatal
Bilateral vestibular schwannomas are diagnostic of NF2.[14]
Postnatal
Ferner et al.[15] give three sets of diagnostic criteria for NF2:
- Bilateral vestibular schwannoma (VS) or family history of NF2 plus Unilateral VS or any two of: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
- Unilateral VS plus any two of meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities
- Two or more meningioma plus unilateral VS or any two of glioma, schwannoma and cataract.
Another set of diagnostic criteria is the following:
- Detection of bilateral acoustic neuroma by imaging-procedures
- First degree relative with NF2 and the occurrence of neurofibroma, meningiomas, glioma, or Schwannoma
- First degree relative with NF2 and the occurrence of juvenile posterior subcapsular cataract.
The criteria have varied over time.[16] The last revision of the NF2 criteria was done by M.J. Smith in 2017. This included the consideration of a LZTR1 mutation (schwannomatosis) instead of NF2 and excluded bilateral vestibular schwannomas that occur after 70 years of age.[17]
Treatment
Surgery
There are several different surgical techniques for the removal of acoustic neuroma.[18] The choice of approach is determined by size of the tumour, hearing capability, and general clinical condition of the person.
- The retrosigmoid approach offers some opportunity for the retention of hearing.
- The translabyrinthine approach will sacrifice hearing on that side, but will usually spare the facial nerve. Post-operative cerebrospinal fluid leaks are more common.
- The middle fossa approach is preferred for small tumors and offers the highest probability of retention of hearing and vestibular function.
- Less invasive endoscopic techniques have been done outside of the United States for some time. Recovery times are reported to be faster. However, this technique is not yet mainstream among surgeons in the US.
Larger tumors can be treated by either the translabyrinthine approach or the retrosigmoid approach, depending upon the experience of the surgical team. With large tumors, the chance of hearing preservation is small with any approach. When hearing is already poor, the translabyrinthine approach may be used for even small tumors. Small, lateralized tumours in people with NF2 with good hearing should have the middle fossa approach. When the location of the tumour is more medial a retrosigmoid approach may be better.
Auditory canal decompression is another surgical technique that can prolong usable hearing when a vestibular schwannoma has grown too large to remove without damage to the cochlear nerve. In the IAC (internal auditory canal) decompression, a middle fossa approach is employed to expose the bony roof of the IAC without any attempt to remove the tumor. The bone overlying the acoustic nerve is removed, allowing the tumour to expand upward into the middle cranial fossa. In this way, pressure on the cochlear nerve is relieved, reducing the risk of further hearing loss from direct compression or obstruction of vascular supply to the nerve.
Radiosurgery is a conservative alternative to cranial base or other intracranial surgery. With conformal radiosurgical techniques, therapeutic radiation focused on the tumour, sparing exposure to surrounding normal tissues. Although radiosurgery can seldom completely destroy a tumor, it can often arrest its growth or reduce its size. While radiation is less immediately damaging than conventional surgery, it incurs a higher risk of subsequent malignant change in the irradiated tissues, and this risk is higher in NF2 than in sporadic (non-NF2) lesions.
Medications
There are no prescription medicines currently indicated for reduction in tumor burden for NF2 patients, although in patient studies Bevacizumab has resulted in reduction in tumor growth rates and hearing improvements in some patients.[19][20]
Hearing loss
Because hearing loss in those with NF2 almost always occurs after acquisition of verbal language skills, people with NF2 do not always integrate well into Deaf culture and are more likely to resort to auditory assistive technology. One of these devices is the cochlear implant, which can sometimes restore a high level of auditory function even when natural hearing is totally lost. However, the amount of destruction to the cochlear nerve caused by the typical NF2 schwannoma often precludes the use of such an implant. In these cases, an auditory brainstem implant (ABI) can restore some level of hearing, supplemented by lip reading.
Prevalence
Incidence of the condition is about 1 in 60,000.[21]
References
- ↑ Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A (November 2008). "The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP". Neoplasia. 10 (11): 1204–12. doi:10.1593/neo.08642. PMC 2570596. PMID 18953429.
- ↑ Asthagiri A, Parry D, Butman J, Kim J, Tsilou E, Zhuang Z, MD, Lonser R (6 June 2009). "Neurofibromatosis type 2". The Lancet. 373 (9679): 1974–1986. doi:10.1016/S0140-6736(09)60259-2. PMC 4748851. PMID 19476995. Archived from the original on 14 June 2020. Retrieved 19 July 2021.
- ↑ Walter J, Kuhn SA, Brodhun M, Reichart R, Kalff R (June 2009). "Pulmonary meningioma and neurinoma associated with multiple CNS tumours in a patient with neurofibromatosis type 2". Clin Neurol Neurosurg. 111 (5): 454–9. doi:10.1016/j.clineuro.2008.11.018. PMID 19249154. S2CID 22696343.
- ↑ "Neurofibromatosis type 2 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 18 March 2021. Retrieved 18 August 2021.
- 1 2 3 Stamenkovic, I; Yu, Q (2010). "Merlin, a "magic" linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival". Current Protein & Peptide Science. 11 (6): 471–84. doi:10.2174/138920310791824011. PMC 2946555. PMID 20491622.
- ↑ Cooper, Jonathan; Giancotti, Filippo G. (2014-04-12). "Molecular insights into NF2/Merlin tumor suppressor function". FEBS Letters. 588 (16): 2743–2752. doi:10.1016/j.febslet.2014.04.001. PMC 4111995. PMID 24726726.
- ↑ Okada, Tomoyo; You, Liru; Giancotti, Filippo G. (May 2007). "Shedding light on Merlin's wizardry". Trends in Cell Biology. 17 (5): 222–229. doi:10.1016/j.tcb.2007.03.006. ISSN 1879-3088. PMID 17442573.
- ↑ Sherman, Larry; Gutmann, David (1 November 2001). "Merlin: hanging tumor suppression on the Rac" (PDF). Trends in Cell Biology. 11 (11): 442–444. doi:10.1016/S0962-8924(01)02128-6. ISSN 0962-8924. PMID 11684412. Archived from the original on 25 April 2021. Retrieved 18 August 2021.
- ↑ Xiao, Guang-Hui; Gallagher, Ryan; Shetler, Justin; Skele, Kristine; Altomare, Deborah; Pestell, Richard; Jhanwar, Suresh; Testa, Joseph (March 2005). "The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression". Molecular and Cellular Biology. 25 (6): 2384–2394. doi:10.1128/MCB.25.6.2384-2394.2005. ISSN 0270-7306. PMC 1061616. PMID 15743831.
- ↑ Cooper, Jonathan; Li, Wei; You, Liru; Schiavon, Gaia; Pepe-Caprio, Angela; Zhou, Lu; Ishii, Ryohei; Giovannini, Marco; Hanemann, C. Oliver (2011-08-23). "Merlin/NF2 functions upstream of the nuclear E3 ubiquitin ligase CRL4DCAF1 to suppress oncogenic gene expression". Science Signaling. 4 (188): pt6. doi:10.1126/scisignal.2002314. ISSN 1937-9145. PMID 21878678. S2CID 9582719.
- ↑ Stepanova, Dina; Braun, Lanita; Chernoff, Jonathan (May 2018). "A new concept in NF2 pharmacotherapy: targeting fatty acid synthesis". Oncoscience. 5 (5–6): 126–127. doi:10.18632/oncoscience.417. ISSN 2331-4737. PMC 6049319. PMID 30035161.
- ↑ Stepanova, Dina; Semenova, Galina; Kuo, Yin-Ming; Andrews, Andrew J.; Ammoun, Sylwia; Hanemann, C. Oliver; Chernoff, Jonathan (15 September 2017). "An Essential Role for the Tumor-Suppressor Merlin in Regulating Fatty Acid Synthesis". Cancer Research. 77 (18): 5026–5038. doi:10.1158/0008-5472.CAN-16-2834. ISSN 1538-7445. PMC 5600854. PMID 28729415.
- ↑ "Ependymoma". The Lecturio Medical Concept Library. Archived from the original on 19 July 2021. Retrieved 19 July 2021.
- ↑ Christopher Gillberg (16 October 2003). Clinical Child Neuropsychiatry. Cambridge University Press. pp. 231–. ISBN 978-0-521-54335-4. Archived from the original on 1 October 2013. Retrieved 20 December 2010.
- ↑ Ferner, Rosalie E., Susan M. Huson, and D. Gareth R. Evans. Neurofibromatoses in clinical practice. Springer, 2011.
- ↑ "Neurofibromatosis Type 2: eMedicine Radiology". 2016-09-26. Archived from the original on 2021-04-25. Retrieved 2010-12-20.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Jean Régis; Pierre-Hugues Roche (2008). Modern Management of Acoustic Neuroma. Karger Publishers. pp. 191–. ISBN 978-3-8055-8370-1. Archived from the original on 3 January 2014. Retrieved 20 December 2010.
- ↑ "Neurofibromatosis Information Page". National Institute of Neurological Disorders and Stroke. Archived from the original on August 16, 2019. Retrieved June 16, 2019.
- ↑ Katrina A. Morris; John F. Golding; Patrick R. Axon; et al. (2016). "Bevacizumab in neurofibromatosis type 2 (NF2) related vestibular schwannomas: a nationally coordinated approach to delivery and prospective evaluation". Neurooncol Pract. 3 (4): 281–289. doi:10.1093/nop/npv065. PMC 5909937. PMID 29692918.
- ↑ Evans DG (2009). "Neurofibromatosis type 2 (NF2): a clinical and molecular review". Orphanet J Rare Dis. 4: 16. doi:10.1186/1750-1172-4-16. PMC 2708144. PMID 19545378.
Further reading
- Evans, D. Gareth (1993). "Neurofibromatosis 2". GeneReviews. University of Washington, Seattle. Archived from the original on 28 January 2017. Retrieved 30 May 2017.
External links
| Classification | |
|---|---|
| External resources |
|