Pharmacodynamics of estradiol
The pharmacology of estradiol, an estrogen medication and naturally occurring steroid hormone, concerns its pharmacodynamics, pharmacokinetics, and various routes of administration.[1][2][3]
Estradiol is a naturally occurring and bioidentical estrogen, or an agonist of the estrogen receptor, the biological target of estrogens like endogenous estradiol.[1] Due to its estrogenic activity, estradiol has antigonadotropic effects and can inhibit fertility and suppress sex hormone production in both women and men.[4][5] Estradiol differs from non-bioidentical estrogens like conjugated estrogens and ethinylestradiol in various ways, with implications for tolerability and safety.[1]
Estradiol can be taken by mouth, held under the tongue, as a gel or patch that is applied to the skin, in through the vagina, by injection into muscle or fat, or through the use of an implant that is placed into fat, among other routes.[1]
Mechanism of action
Estradiol is an estrogen, or an agonist of the nuclear estrogen receptors (ERs), the estrogen receptor alpha (ERα) and the estrogen receptor beta (ERβ).[1][2][6] In one study, the EC50 value of estradiol for the human ERα was 50 pM (0.05 nM) and for the human ERβ was 200 pM (0.2 nM).[2][7] Estradiol is also an agonist of the membrane estrogen receptors (mERs), including the G protein-coupled estrogen receptor (GPER) (3–6 nM),[8] Gq-coupled membrane estrogen receptor (Gq-mER), ER-X, and ERx.[9][10] It is far more potent as an estrogen than are other natural and bioidentical estrogens like estrone and estriol.[1] Given by subcutaneous injection in mice, estradiol is about 10-fold more potent than estrone and about 100-fold more potent than estriol.[11] In addition, much of the estrogenic potency of estrone in vivo is actually due to conversion into estradiol.[1]
Estradiol has little to no affinity for other steroid hormone receptors, including the androgen, progesterone, glucocorticoid, and mineralocorticoid receptors.[12][13][14] It has weak affinity for the androgen receptor, with about 8% of relative binding affinity of testosterone according to one study,[15] and shows agonistic activity at this receptor.[16] However, estrogens circulate in the picomolar (10−12 M) range while androgens circulate in the nanomolar (10−9 M) to micromolar (10−6 M) range,[17][18] and in accordance with this, estradiol is active as an estrogen in target tissues at approximately 1,000-fold lower concentrations than is testosterone.[19] In addition, while estradiol did show activation of the androgen receptor in vitro at very high concentrations, its efficacy as an androgen receptor agonist was of such low potency that it was not possible to calculate an EC50 value for the activity.[16] As such, the weak activity of estradiol at the androgen receptor is unlikely to be of biological significance at normal physiological concentrations.[15][16]
The affinities of estradiol for the ERs are high (around 0.1 nM), and there is a relatively low quantity of about 10,000 to 20,000 ERs in the cytoplasm per cell in estrogen target tissues.[20] Estradiol stays bound to the ERs for about 24 hours, which is longer than that of other estrogens such as estriol (6 hours).[1] A prolonged duration of binding to the ERs (e.g., 9 to 12 hours for endometrial effects), as with estradiol, is necessary for full estrogenic responses in various tissues.[1] The ERs downregulate with exposure to estradiol, and in accordance, the expression of the ERs is dependent on estradiol concentrations.[21][22] Constant levels of estradiol may result in downregulation of the ERs and relatively diminished responses to estradiol, although this has not been assessed clinically.[21] Once bound to estradiol, the ERs are ubiquitinated and degraded by proteasomes, which is a major mechanism of ER downregulation.[22] The unbound ERα has an intracellular half-life of up to 5 days, but this shortens to 3–4 hours once bound to a ligand such as estradiol.[23][22] Estrogen deprivation can easily increase sensitivity to estrogens like estradiol by 10,000-fold or more, demonstrating a profound capacity of the ERs for upregulation and downregulation.[24] This increase in sensitivity is mediated by a 100-fold increase in ERs, as well as other mechanisms such as changes in coactivator sensitivity and degree of phosphorylation of transactivation factors.[24] Progestogens like progesterone and androgens like testosterone downregulate the ERs in certain tissues such as the endometrium and breasts, among others.[25][1][26]
Estradiol is a steroid and a lipophilic compound.[1][27] As a result, it readily enters cells via simple passive diffusion through the lipid bilayer of the cell membrane.[27] This is in contrast to hydrophilic estrogen conjugates such as estrone sulfate and estradiol glucuronide, which require active transport via specific membrane transport proteins to enter cells.[28][29][30] The ERs are nuclear receptors that are mostly present in the cell nucleus.[27] Upon binding of estradiol to an ER, the receptor dimerizes (combines) with another estradiol-bound ER.[1][27] These ER dimers can be ERα–ERα or ERβ–ERβ homodimers or ERα–ERβ heterodimers.[1] Once in the dimerized state, the estradiol-bound ER–ER complex binds to short estrogen response elements (EREs) (of the minimal nucleotide sequence 5'-GGTCANNNTGACC-3', where N is any nucleotide) in the promoter regions of estrogen-responsive genes on chromosomes, in turn modulating their expression.[1][27][31] Some prominent examples ERE-containing and hence estrogen-modulated genes in humans include the genes encoding the proteins oxytocin, c-fos, c-myc, and transforming growth factor alpha (TGFα).[32]
| Ligand | Other names | Relative binding affinities (RBA, %)a | Absolute binding affinities (Ki, nM)a | Action | ||
|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ERβ | |||
| Estradiol | E2; 17β-Estradiol | 100 | 100 | 0.115 (0.04–0.24) | 0.15 (0.10–2.08) | Estrogen |
| Estrone | E1; 17-Ketoestradiol | 16.39 (0.7–60) | 6.5 (1.36–52) | 0.445 (0.3–1.01) | 1.75 (0.35–9.24) | Estrogen |
| Estriol | E3; 16α-OH-17β-E2 | 12.65 (4.03–56) | 26 (14.0–44.6) | 0.45 (0.35–1.4) | 0.7 (0.63–0.7) | Estrogen |
| Estetrol | E4; 15α,16α-Di-OH-17β-E2 | 4.0 | 3.0 | 4.9 | 19 | Estrogen |
| Alfatradiol | 17α-Estradiol | 20.5 (7–80.1) | 8.195 (2–42) | 0.2–0.52 | 0.43–1.2 | Metabolite |
| 16-Epiestriol | 16β-Hydroxy-17β-estradiol | 7.795 (4.94–63) | 50 | ? | ? | Metabolite |
| 17-Epiestriol | 16α-Hydroxy-17α-estradiol | 55.45 (29–103) | 79–80 | ? | ? | Metabolite |
| 16,17-Epiestriol | 16β-Hydroxy-17α-estradiol | 1.0 | 13 | ? | ? | Metabolite |
| 2-Hydroxyestradiol | 2-OH-E2 | 22 (7–81) | 11–35 | 2.5 | 1.3 | Metabolite |
| 2-Methoxyestradiol | 2-MeO-E2 | 0.0027–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Hydroxyestradiol | 4-OH-E2 | 13 (8–70) | 7–56 | 1.0 | 1.9 | Metabolite |
| 4-Methoxyestradiol | 4-MeO-E2 | 2.0 | 1.0 | ? | ? | Metabolite |
| 2-Hydroxyestrone | 2-OH-E1 | 2.0–4.0 | 0.2–0.4 | ? | ? | Metabolite |
| 2-Methoxyestrone | 2-MeO-E1 | <0.001–<1 | <1 | ? | ? | Metabolite |
| 4-Hydroxyestrone | 4-OH-E1 | 1.0–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestrone | 4-MeO-E1 | <1 | <1 | ? | ? | Metabolite |
| 16α-Hydroxyestrone | 16α-OH-E1; 17-Ketoestriol | 2.0–6.5 | 35 | ? | ? | Metabolite |
| 2-Hydroxyestriol | 2-OH-E3 | 2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestriol | 4-MeO-E3 | 1.0 | 1.0 | ? | ? | Metabolite |
| Estradiol sulfate | E2S; Estradiol 3-sulfate | <1 | <1 | ? | ? | Metabolite |
| Estradiol disulfate | Estradiol 3,17β-disulfate | 0.0004 | ? | ? | ? | Metabolite |
| Estradiol 3-glucuronide | E2-3G | 0.0079 | ? | ? | ? | Metabolite |
| Estradiol 17β-glucuronide | E2-17G | 0.0015 | ? | ? | ? | Metabolite |
| Estradiol 3-gluc. 17β-sulfate | E2-3G-17S | 0.0001 | ? | ? | ? | Metabolite |
| Estrone sulfate | E1S; Estrone 3-sulfate | <1 | <1 | >10 | >10 | Metabolite |
| Estradiol benzoate | EB; Estradiol 3-benzoate | 10 | ? | ? | ? | Estrogen |
| Estradiol 17β-benzoate | E2-17B | 11.3 | 32.6 | ? | ? | Estrogen |
| Estrone methyl ether | Estrone 3-methyl ether | 0.145 | ? | ? | ? | Estrogen |
| ent-Estradiol | 1-Estradiol | 1.31–12.34 | 9.44–80.07 | ? | ? | Estrogen |
| Equilin | 7-Dehydroestrone | 13 (4.0–28.9) | 13.0–49 | 0.79 | 0.36 | Estrogen |
| Equilenin | 6,8-Didehydroestrone | 2.0–15 | 7.0–20 | 0.64 | 0.62 | Estrogen |
| 17β-Dihydroequilin | 7-Dehydro-17β-estradiol | 7.9–113 | 7.9–108 | 0.09 | 0.17 | Estrogen |
| 17α-Dihydroequilin | 7-Dehydro-17α-estradiol | 18.6 (18–41) | 14–32 | 0.24 | 0.57 | Estrogen |
| 17β-Dihydroequilenin | 6,8-Didehydro-17β-estradiol | 35–68 | 90–100 | 0.15 | 0.20 | Estrogen |
| 17α-Dihydroequilenin | 6,8-Didehydro-17α-estradiol | 20 | 49 | 0.50 | 0.37 | Estrogen |
| Δ8-Estradiol | 8,9-Dehydro-17β-estradiol | 68 | 72 | 0.15 | 0.25 | Estrogen |
| Δ8-Estrone | 8,9-Dehydroestrone | 19 | 32 | 0.52 | 0.57 | Estrogen |
| Ethinylestradiol | EE; 17α-Ethynyl-17β-E2 | 120.9 (68.8–480) | 44.4 (2.0–144) | 0.02–0.05 | 0.29–0.81 | Estrogen |
| Mestranol | EE 3-methyl ether | ? | 2.5 | ? | ? | Estrogen |
| Moxestrol | RU-2858; 11β-Methoxy-EE | 35–43 | 5–20 | 0.5 | 2.6 | Estrogen |
| Methylestradiol | 17α-Methyl-17β-estradiol | 70 | 44 | ? | ? | Estrogen |
| Diethylstilbestrol | DES; Stilbestrol | 129.5 (89.1–468) | 219.63 (61.2–295) | 0.04 | 0.05 | Estrogen |
| Hexestrol | Dihydrodiethylstilbestrol | 153.6 (31–302) | 60–234 | 0.06 | 0.06 | Estrogen |
| Dienestrol | Dehydrostilbestrol | 37 (20.4–223) | 56–404 | 0.05 | 0.03 | Estrogen |
| Benzestrol (B2) | – | 114 | ? | ? | ? | Estrogen |
| Chlorotrianisene | TACE | 1.74 | ? | 15.30 | ? | Estrogen |
| Triphenylethylene | TPE | 0.074 | ? | ? | ? | Estrogen |
| Triphenylbromoethylene | TPBE | 2.69 | ? | ? | ? | Estrogen |
| Tamoxifen | ICI-46,474 | 3 (0.1–47) | 3.33 (0.28–6) | 3.4–9.69 | 2.5 | SERM |
| Afimoxifene | 4-Hydroxytamoxifen; 4-OHT | 100.1 (1.7–257) | 10 (0.98–339) | 2.3 (0.1–3.61) | 0.04–4.8 | SERM |
| Toremifene | 4-Chlorotamoxifen; 4-CT | ? | ? | 7.14–20.3 | 15.4 | SERM |
| Clomifene | MRL-41 | 25 (19.2–37.2) | 12 | 0.9 | 1.2 | SERM |
| Cyclofenil | F-6066; Sexovid | 151–152 | 243 | ? | ? | SERM |
| Nafoxidine | U-11,000A | 30.9–44 | 16 | 0.3 | 0.8 | SERM |
| Raloxifene | – | 41.2 (7.8–69) | 5.34 (0.54–16) | 0.188–0.52 | 20.2 | SERM |
| Arzoxifene | LY-353,381 | ? | ? | 0.179 | ? | SERM |
| Lasofoxifene | CP-336,156 | 10.2–166 | 19.0 | 0.229 | ? | SERM |
| Ormeloxifene | Centchroman | ? | ? | 0.313 | ? | SERM |
| Levormeloxifene | 6720-CDRI; NNC-460,020 | 1.55 | 1.88 | ? | ? | SERM |
| Ospemifene | Deaminohydroxytoremifene | 0.82–2.63 | 0.59–1.22 | ? | ? | SERM |
| Bazedoxifene | – | ? | ? | 0.053 | ? | SERM |
| Etacstil | GW-5638 | 4.30 | 11.5 | ? | ? | SERM |
| ICI-164,384 | – | 63.5 (3.70–97.7) | 166 | 0.2 | 0.08 | Antiestrogen |
| Fulvestrant | ICI-182,780 | 43.5 (9.4–325) | 21.65 (2.05–40.5) | 0.42 | 1.3 | Antiestrogen |
| Propylpyrazoletriol | PPT | 49 (10.0–89.1) | 0.12 | 0.40 | 92.8 | ERα agonist |
| 16α-LE2 | 16α-Lactone-17β-estradiol | 14.6–57 | 0.089 | 0.27 | 131 | ERα agonist |
| 16α-Iodo-E2 | 16α-Iodo-17β-estradiol | 30.2 | 2.30 | ? | ? | ERα agonist |
| Methylpiperidinopyrazole | MPP | 11 | 0.05 | ? | ? | ERα antagonist |
| Diarylpropionitrile | DPN | 0.12–0.25 | 6.6–18 | 32.4 | 1.7 | ERβ agonist |
| 8β-VE2 | 8β-Vinyl-17β-estradiol | 0.35 | 22.0–83 | 12.9 | 0.50 | ERβ agonist |
| Prinaberel | ERB-041; WAY-202,041 | 0.27 | 67–72 | ? | ? | ERβ agonist |
| ERB-196 | WAY-202,196 | ? | 180 | ? | ? | ERβ agonist |
| Erteberel | SERBA-1; LY-500,307 | ? | ? | 2.68 | 0.19 | ERβ agonist |
| SERBA-2 | – | ? | ? | 14.5 | 1.54 | ERβ agonist |
| Coumestrol | – | 9.225 (0.0117–94) | 64.125 (0.41–185) | 0.14–80.0 | 0.07–27.0 | Xenoestrogen |
| Genistein | – | 0.445 (0.0012–16) | 33.42 (0.86–87) | 2.6–126 | 0.3–12.8 | Xenoestrogen |
| Equol | – | 0.2–0.287 | 0.85 (0.10–2.85) | ? | ? | Xenoestrogen |
| Daidzein | – | 0.07 (0.0018–9.3) | 0.7865 (0.04–17.1) | 2.0 | 85.3 | Xenoestrogen |
| Biochanin A | – | 0.04 (0.022–0.15) | 0.6225 (0.010–1.2) | 174 | 8.9 | Xenoestrogen |
| Kaempferol | – | 0.07 (0.029–0.10) | 2.2 (0.002–3.00) | ? | ? | Xenoestrogen |
| Naringenin | – | 0.0054 (<0.001–0.01) | 0.15 (0.11–0.33) | ? | ? | Xenoestrogen |
| 8-Prenylnaringenin | 8-PN | 4.4 | ? | ? | ? | Xenoestrogen |
| Quercetin | – | <0.001–0.01 | 0.002–0.040 | ? | ? | Xenoestrogen |
| Ipriflavone | – | <0.01 | <0.01 | ? | ? | Xenoestrogen |
| Miroestrol | – | 0.39 | ? | ? | ? | Xenoestrogen |
| Deoxymiroestrol | – | 2.0 | ? | ? | ? | Xenoestrogen |
| β-Sitosterol | – | <0.001–0.0875 | <0.001–0.016 | ? | ? | Xenoestrogen |
| Resveratrol | – | <0.001–0.0032 | ? | ? | ? | Xenoestrogen |
| α-Zearalenol | – | 48 (13–52.5) | ? | ? | ? | Xenoestrogen |
| β-Zearalenol | – | 0.6 (0.032–13) | ? | ? | ? | Xenoestrogen |
| Zeranol | α-Zearalanol | 48–111 | ? | ? | ? | Xenoestrogen |
| Taleranol | β-Zearalanol | 16 (13–17.8) | 14 | 0.8 | 0.9 | Xenoestrogen |
| Zearalenone | ZEN | 7.68 (2.04–28) | 9.45 (2.43–31.5) | ? | ? | Xenoestrogen |
| Zearalanone | ZAN | 0.51 | ? | ? | ? | Xenoestrogen |
| Bisphenol A | BPA | 0.0315 (0.008–1.0) | 0.135 (0.002–4.23) | 195 | 35 | Xenoestrogen |
| Endosulfan | EDS | <0.001–<0.01 | <0.01 | ? | ? | Xenoestrogen |
| Kepone | Chlordecone | 0.0069–0.2 | ? | ? | ? | Xenoestrogen |
| o,p'-DDT | – | 0.0073–0.4 | ? | ? | ? | Xenoestrogen |
| p,p'-DDT | – | 0.03 | ? | ? | ? | Xenoestrogen |
| Methoxychlor | p,p'-Dimethoxy-DDT | 0.01 (<0.001–0.02) | 0.01–0.13 | ? | ? | Xenoestrogen |
| HPTE | Hydroxychlor; p,p'-OH-DDT | 1.2–1.7 | ? | ? | ? | Xenoestrogen |
| Testosterone | T; 4-Androstenolone | <0.0001–<0.01 | <0.002–0.040 | >5000 | >5000 | Androgen |
| Dihydrotestosterone | DHT; 5α-Androstanolone | 0.01 (<0.001–0.05) | 0.0059–0.17 | 221–>5000 | 73–1688 | Androgen |
| Nandrolone | 19-Nortestosterone; 19-NT | 0.01 | 0.23 | 765 | 53 | Androgen |
| Dehydroepiandrosterone | DHEA; Prasterone | 0.038 (<0.001–0.04) | 0.019–0.07 | 245–1053 | 163–515 | Androgen |
| 5-Androstenediol | A5; Androstenediol | 6 | 17 | 3.6 | 0.9 | Androgen |
| 4-Androstenediol | – | 0.5 | 0.6 | 23 | 19 | Androgen |
| 4-Androstenedione | A4; Androstenedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| 3α-Androstanediol | 3α-Adiol | 0.07 | 0.3 | 260 | 48 | Androgen |
| 3β-Androstanediol | 3β-Adiol | 3 | 7 | 6 | 2 | Androgen |
| Androstanedione | 5α-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Etiocholanedione | 5β-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Methyltestosterone | 17α-Methyltestosterone | <0.0001 | ? | ? | ? | Androgen |
| Ethinyl-3α-androstanediol | 17α-Ethynyl-3α-adiol | 4.0 | <0.07 | ? | ? | Estrogen |
| Ethinyl-3β-androstanediol | 17α-Ethynyl-3β-adiol | 50 | 5.6 | ? | ? | Estrogen |
| Progesterone | P4; 4-Pregnenedione | <0.001–0.6 | <0.001–0.010 | ? | ? | Progestogen |
| Norethisterone | NET; 17α-Ethynyl-19-NT | 0.085 (0.0015–<0.1) | 0.1 (0.01–0.3) | 152 | 1084 | Progestogen |
| Norethynodrel | 5(10)-Norethisterone | 0.5 (0.3–0.7) | <0.1–0.22 | 14 | 53 | Progestogen |
| Tibolone | 7α-Methylnorethynodrel | 0.5 (0.45–2.0) | 0.2–0.076 | ? | ? | Progestogen |
| Δ4-Tibolone | 7α-Methylnorethisterone | 0.069–<0.1 | 0.027–<0.1 | ? | ? | Progestogen |
| 3α-Hydroxytibolone | – | 2.5 (1.06–5.0) | 0.6–0.8 | ? | ? | Progestogen |
| 3β-Hydroxytibolone | – | 1.6 (0.75–1.9) | 0.070–0.1 | ? | ? | Progestogen |
| Footnotes: a = (1) Binding affinity values are of the format "median (range)" (# (#–#)), "range" (#–#), or "value" (#) depending on the values available. The full sets of values within the ranges can be found in the Wiki code. (2) Binding affinities were determined via displacement studies in a variety of in-vitro systems with labeled estradiol and human ERα and ERβ proteins (except the ERβ values from Kuiper et al. (1997), which are rat ERβ). Sources: See template page. | ||||||
| Estrogen | Relative binding affinities (%) | ||||||
|---|---|---|---|---|---|---|---|
| ER | AR | PR | GR | MR | SHBG | CBG | |
| Estradiol | 100 | 7.9 | 2.6 | 0.6 | 0.13 | 8.7–12 | <0.1 |
| Estradiol benzoate | ? | ? | ? | ? | ? | <0.1–0.16 | <0.1 |
| Estradiol valerate | 2 | ? | ? | ? | ? | ? | ? |
| Estrone | 11–35 | <1 | <1 | <1 | <1 | 2.7 | <0.1 |
| Estrone sulfate | 2 | 2 | ? | ? | ? | ? | ? |
| Estriol | 10–15 | <1 | <1 | <1 | <1 | <0.1 | <0.1 |
| Equilin | 40 | ? | ? | ? | ? | ? | 0 |
| Alfatradiol | 15 | <1 | <1 | <1 | <1 | ? | ? |
| Epiestriol | 20 | <1 | <1 | <1 | <1 | ? | ? |
| Ethinylestradiol | 100–112 | 1–3 | 15–25 | 1–3 | <1 | 0.18 | <0.1 |
| Mestranol | 1 | ? | ? | ? | ? | <0.1 | <0.1 |
| Methylestradiol | 67 | 1–3 | 3–25 | 1–3 | <1 | ? | ? |
| Moxestrol | 12 | <0.1 | 0.8 | 3.2 | <0.1 | <0.2 | <0.1 |
| Diethylstilbestrol | ? | ? | ? | ? | ? | <0.1 | <0.1 |
| Notes: Reference ligands (100%) were progesterone for the PR, testosterone for the AR, estradiol for the ER, dexamethasone for the GR, aldosterone for the MR, dihydrotestosterone for SHBG, and cortisol for CBG. Sources: See template. | |||||||
| Estrogen | Other names | RBA (%)a | REP (%)b | |||
|---|---|---|---|---|---|---|
| ER | ERα | ERβ | ||||
| Estradiol | E2 | 100 | 100 | 100 | ||
| Estradiol 3-sulfate | E2S; E2-3S | ? | 0.02 | 0.04 | ||
| Estradiol 3-glucuronide | E2-3G | ? | 0.02 | 0.09 | ||
| Estradiol 17β-glucuronide | E2-17G | ? | 0.002 | 0.0002 | ||
| Estradiol benzoate | EB; Estradiol 3-benzoate | 10 | 1.1 | 0.52 | ||
| Estradiol 17β-acetate | E2-17A | 31–45 | 24 | ? | ||
| Estradiol diacetate | EDA; Estradiol 3,17β-diacetate | ? | 0.79 | ? | ||
| Estradiol propionate | EP; Estradiol 17β-propionate | 19–26 | 2.6 | ? | ||
| Estradiol valerate | EV; Estradiol 17β-valerate | 2–11 | 0.04–21 | ? | ||
| Estradiol cypionate | EC; Estradiol 17β-cypionate | ?c | 4.0 | ? | ||
| Estradiol palmitate | Estradiol 17β-palmitate | 0 | ? | ? | ||
| Estradiol stearate | Estradiol 17β-stearate | 0 | ? | ? | ||
| Estrone | E1; 17-Ketoestradiol | 11 | 5.3–38 | 14 | ||
| Estrone sulfate | E1S; Estrone 3-sulfate | 2 | 0.004 | 0.002 | ||
| Estrone glucuronide | E1G; Estrone 3-glucuronide | ? | <0.001 | 0.0006 | ||
| Ethinylestradiol | EE; 17α-Ethynylestradiol | 100 | 17–150 | 129 | ||
| Mestranol | EE 3-methyl ether | 1 | 1.3–8.2 | 0.16 | ||
| Quinestrol | EE 3-cyclopentyl ether | ? | 0.37 | ? | ||
| Footnotes: a = Relative binding affinities (RBAs) were determined via in-vitro displacement of labeled estradiol from estrogen receptors (ERs) generally of rodent uterine cytosol. Estrogen esters are variably hydrolyzed into estrogens in these systems (shorter ester chain length -> greater rate of hydrolysis) and the ER RBAs of the esters decrease strongly when hydrolysis is prevented. b = Relative estrogenic potencies (REPs) were calculated from half-maximal effective concentrations (EC50) that were determined via in-vitro β‐galactosidase (β-gal) and green fluorescent protein (GFP) production assays in yeast expressing human ERα and human ERβ. Both mammalian cells and yeast have the capacity to hydrolyze estrogen esters. c = The affinities of estradiol cypionate for the ERs are similar to those of estradiol valerate and estradiol benzoate (figure). Sources: See template page. | ||||||
| Estrogen | ER RBA (%) | Uterine weight (%) | Uterotrophy | LH levels (%) | SHBG RBA (%) |
|---|---|---|---|---|---|
| Control | – | 100 | – | 100 | – |
| Estradiol (E2) | 100 | 506 ± 20 | +++ | 12–19 | 100 |
| Estrone (E1) | 11 ± 8 | 490 ± 22 | +++ | ? | 20 |
| Estriol (E3) | 10 ± 4 | 468 ± 30 | +++ | 8–18 | 3 |
| Estetrol (E4) | 0.5 ± 0.2 | ? | Inactive | ? | 1 |
| 17α-Estradiol | 4.2 ± 0.8 | ? | ? | ? | ? |
| 2-Hydroxyestradiol | 24 ± 7 | 285 ± 8 | +b | 31–61 | 28 |
| 2-Methoxyestradiol | 0.05 ± 0.04 | 101 | Inactive | ? | 130 |
| 4-Hydroxyestradiol | 45 ± 12 | ? | ? | ? | ? |
| 4-Methoxyestradiol | 1.3 ± 0.2 | 260 | ++ | ? | 9 |
| 4-Fluoroestradiola | 180 ± 43 | ? | +++ | ? | ? |
| 2-Hydroxyestrone | 1.9 ± 0.8 | 130 ± 9 | Inactive | 110–142 | 8 |
| 2-Methoxyestrone | 0.01 ± 0.00 | 103 ± 7 | Inactive | 95–100 | 120 |
| 4-Hydroxyestrone | 11 ± 4 | 351 | ++ | 21–50 | 35 |
| 4-Methoxyestrone | 0.13 ± 0.04 | 338 | ++ | 65–92 | 12 |
| 16α-Hydroxyestrone | 2.8 ± 1.0 | 552 ± 42 | +++ | 7–24 | <0.5 |
| 2-Hydroxyestriol | 0.9 ± 0.3 | 302 | +b | ? | ? |
| 2-Methoxyestriol | 0.01 ± 0.00 | ? | Inactive | ? | 4 |
| Notes: Values are mean ± SD or range. ER RBA = Relative binding affinity to estrogen receptors of rat uterine cytosol. Uterine weight = Percentage change in uterine wet weight of ovariectomized rats after 72 hours with continuous administration of 1 μg/hour via subcutaneously implanted osmotic pumps. LH levels = Luteinizing hormone levels relative to baseline of ovariectomized rats after 24 to 72 hours of continuous administration via subcutaneous implant. Footnotes: a = Synthetic (i.e., not endogenous). b = Atypical uterotrophic effect which plateaus within 48 hours (estradiol's uterotrophy continues linearly up to 72 hours). Sources: See template. | |||||
Effects in the body and brain
The ERs are expressed widely throughout the body, including in the breasts, uterus, vagina, prostate gland, fat, skin, bone, liver, pituitary gland, hypothalamus, and elsewhere throughout the brain.[33] Through activation of the ERs (as well as the mERs), estradiol has many effects, including the following:
- Promotes growth, function, and maintenance of the breasts, uterus, and vagina during puberty and thereafter[33][34]
- Mediates deposition of subcutaneous fat in a feminine pattern, especially in the breasts, hips, buttocks, and thighs[35]
- Maintains skin health, integrity, appearance, and hydration and slows the rate of aging of the skin[36]
- Produces the growth spurt and epiphyseal closure in both sexes during puberty, mediates widening of the hips in females during puberty, and maintains bone mineral density in both sexes throughout life[37][38]
- Modulates hepatic protein synthesis, such as the production of sex hormone-binding globulin (SHBG) and numerous other proteins, with consequent effects on the cardiovascular system and various other systems[3]
- Exerts negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis) by suppressing the secretion of the gonadotropins FSH and LH from the pituitary gland, thereby inhibiting gonadal sex hormone production as well as ovulation and fertility[39][3][40]
- Regulates the vasomotor system and body temperature via the hypothalamus, thereby preventing hot flashes[41][42]
- Modulates brain function, with effects on mood, emotionality, and sexuality, as well as cognition and memory[43]
- Influences the risk and/or progression of hormone-sensitive cancers including breast cancer, prostate cancer, and endometrial cancer[44][3]
Estrogen has also been found to increase the secretion of oxytocin and to increase the expression of its receptor, the oxytocin receptor, in the brain.[18] In women, a single dose of estradiol has been found to be sufficient to increase circulating oxytocin concentrations.[45]
| Compound | Dosage for specific uses (mg usually)[lower-alpha 1] | ||||||
|---|---|---|---|---|---|---|---|
| ETD[lower-alpha 2] | EPD[lower-alpha 2] | MSD[lower-alpha 2] | MSD[lower-alpha 3] | OID[lower-alpha 3] | TSD[lower-alpha 3] | ||
| Estradiol (non-micron.) | 30 | ≥120–300 | 120 | 6 | - | - | |
| Estradiol (micronized) | 6–12 | 60–80 | 14–42 | 1–2 | >5 | >8 | |
| Estradiol valerate | 6–12 | 60–80 | 14–42 | 1–2 | - | >8 | |
| Estradiol benzoate | - | 60–140 | - | - | - | - | |
| Estriol | ≥20 | 120–150[lower-alpha 4] | 28–126 | 1–6 | >5 | - | |
| Estriol succinate | - | 140–150[lower-alpha 4] | 28–126 | 2–6 | - | - | |
| Estrone sulfate | 12 | 60 | 42 | 2 | - | - | |
| Conjugated estrogens | 5–12 | 60–80 | 8.4–25 | 0.625–1.25 | >3.75 | 7.5 | |
| Ethinylestradiol | 200 μg | 1–2 | 280 μg | 20–40 μg | 100 μg | 100 μg | |
| Mestranol | 300 μg | 1.5–3.0 | 300–600 μg | 25–30 μg | >80 μg | - | |
| Quinestrol | 300 μg | 2–4 | 500 μg | 25–50 μg | - | - | |
| Methylestradiol | - | 2 | - | - | - | - | |
| Diethylstilbestrol | 2.5 | 20–30 | 11 | 0.5–2.0 | >5 | 3 | |
| DES dipropionate | - | 15–30 | - | - | - | - | |
| Dienestrol | 5 | 30–40 | 42 | 0.5–4.0 | - | - | |
| Dienestrol diacetate | 3–5 | 30–60 | - | - | - | - | |
| Hexestrol | - | 70–110 | - | - | - | - | |
| Chlorotrianisene | - | >100 | - | - | >48 | - | |
| Methallenestril | - | 400 | - | - | - | - | |
| Estrogen | HF | VE | UCa | FSH | LH | HDL-C | SHBG | CBG | AGT | Liver |
|---|---|---|---|---|---|---|---|---|---|---|
| Estradiol | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Estrone | ? | ? | ? | 0.3 | 0.3 | ? | ? | ? | ? | ? |
| Estriol | 0.3 | 0.3 | 0.1 | 0.3 | 0.3 | 0.2 | ? | ? | ? | 0.67 |
| Estrone sulfate | ? | 0.9 | 0.9 | 0.8–0.9 | 0.9 | 0.5 | 0.9 | 0.5–0.7 | 1.4–1.5 | 0.56–1.7 |
| Conjugated estrogens | 1.2 | 1.5 | 2.0 | 1.1–1.3 | 1.0 | 1.5 | 3.0–3.2 | 1.3–1.5 | 5.0 | 1.3–4.5 |
| Equilin sulfate | ? | ? | 1.0 | ? | ? | 6.0 | 7.5 | 6.0 | 7.5 | ? |
| Ethinylestradiol | 120 | 150 | 400 | 60–150 | 100 | 400 | 500–600 | 500–600 | 350 | 2.9–5.0 |
| Diethylstilbestrol | ? | ? | ? | 2.9–3.4 | ? | ? | 26–28 | 25–37 | 20 | 5.7–7.5 |
Sources and footnotes
Notes: Values are ratios, with estradiol as standard (i.e., 1.0). Abbreviations: HF = Clinical relief of hot flashes. VE = Increased proliferation of vaginal epithelium. UCa = Decrease in UCa. FSH = Suppression of FSH levels. LH = Suppression of LH levels. HDL-C, SHBG, CBG, and AGT = Increase in the serum levels of these liver proteins. Liver = Ratio of liver estrogenic effects to general/systemic estrogenic effects (hot flashes/gonadotropins). Sources: See template. | ||||||||||
| Estrogen | Form | Dose (mg) | Duration by dose (mg) | ||
|---|---|---|---|---|---|
| EPD | CICD | ||||
| Estradiol | Aq. soln. | ? | – | <1 d | |
| Oil soln. | 40–60 | – | 1–2 ≈ 1–2 d | ||
| Aq. susp. | ? | 3.5 | 0.5–2 ≈ 2–7 d; 3.5 ≈ >5 d | ||
| Microsph. | ? | – | 1 ≈ 30 d | ||
| Estradiol benzoate | Oil soln. | 25–35 | – | 1.66 ≈ 2–3 d; 5 ≈ 3–6 d | |
| Aq. susp. | 20 | – | 10 ≈ 16–21 d | ||
| Emulsion | ? | – | 10 ≈ 14–21 d | ||
| Estradiol dipropionate | Oil soln. | 25–30 | – | 5 ≈ 5–8 d | |
| Estradiol valerate | Oil soln. | 20–30 | 5 | 5 ≈ 7–8 d; 10 ≈ 10–14 d; 40 ≈ 14–21 d; 100 ≈ 21–28 d | |
| Estradiol benz. butyrate | Oil soln. | ? | 10 | 10 ≈ 21 d | |
| Estradiol cypionate | Oil soln. | 20–30 | – | 5 ≈ 11–14 d | |
| Aq. susp. | ? | 5 | 5 ≈ 14–24 d | ||
| Estradiol enanthate | Oil soln. | ? | 5–10 | 10 ≈ 20–30 d | |
| Estradiol dienanthate | Oil soln. | ? | – | 7.5 ≈ >40 d | |
| Estradiol undecylate | Oil soln. | ? | – | 10–20 ≈ 40–60 d; 25–50 ≈ 60–120 d | |
| Polyestradiol phosphate | Aq. soln. | 40–60 | – | 40 ≈ 30 d; 80 ≈ 60 d; 160 ≈ 120 d | |
| Estrone | Oil soln. | ? | – | 1–2 ≈ 2–3 d | |
| Aq. susp. | ? | – | 0.1–2 ≈ 2–7 d | ||
| Estriol | Oil soln. | ? | – | 1–2 ≈ 1–4 d | |
| Polyestriol phosphate | Aq. soln. | ? | – | 50 ≈ 30 d; 80 ≈ 60 d | |
Notes and sources
Notes: All aqueous suspensions are of microcrystalline particle size. Estradiol production during the menstrual cycle is 30–640 µg/d (6.4–8.6 mg total per month or cycle). The vaginal epithelium maturation dosage of estradiol benzoate or estradiol valerate has been reported as 5 to 7 mg/week. An effective ovulation-inhibiting dose of estradiol undecylate is 20–30 mg/month. Sources: See template. | |||||
Antigonadotropic effects
Estrogens are powerful antigonadotropins at sufficiently high concentrations.[40][65][66][4][5] By exerting negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis), they are able to suppress the secretion of the gonadotropins, LH and FSH, and thereby inhibit gonadal sex hormone production and circulating sex hormone levels as well as fertility (ovulation in women and spermatogenesis in men).[40][65][66] Clinical studies have found that in men treated with them, estrogens can maximally suppress testosterone levels by about 95% or well into the castrate/female range (<50 ng/dL).[4][5] This is equivalent to the reduction in testosterone levels achieved by orchiectomy and gonadotropin-releasing hormone analogue (GnRH analogue) therapy, corresponding to a complete shutdown of gonadal testosterone production.[67][68] In addition, it is greater than that achieved with high-dose progestogens like cyproterone acetate and gestonorone caproate, which can maximally suppress testosterone levels in men by about 75%.[69][70][71][72][73]
Inhibition of ovulation by estradiol monotherapy in women has been studied and demonstrated for oral estradiol, transdermal estradiol patches, subcutaneous estradiol implants, and intramuscular estradiol undecylate injections.[74][75][76][77][78][79] A study of ovulation inhibition in women found that oral non-micronized estradiol was 55% effective at 1 mg/day, 61% effective at 2 mg/day, and 88% effective at 5 mg/day.[79][80]
Suppression of testosterone levels by estradiol to within the castrate/female range (<50 ng/dL) in men requires relatively high levels of estradiol and has been associated with circulating levels of 200 to 300 pg/mL and above.[81][5] However, although the castrate range in men has been defined as testosterone concentrations of less than 50 ng/dL, mean levels of testosterone with surgical castration are actually about 15 ng/dL.[82] To achieve such levels of testosterone with estradiol therapy, higher concentrations of estradiol of about 500 pg/mL have been necessary to produce the requisite maximal suppression of testosterone production.[4] Injected estradiol esters like polyestradiol phosphate, estradiol valerate, and estradiol undecylate, as well as high-dose estradiol transdermal patches, are used as a form of high-dose estrogen therapy to suppress testosterone levels into the castrate range in men with prostate cancer.[3][83][84][85][5][71] High dosages of estradiol in various forms and routes are also used to suppress testosterone levels in transgender women.[86][87][88] The suppression of testosterone levels by estradiol in men is rapid.[89] A single intramuscular injection of 2 mg aqueous estradiol suppressed testosterone levels in young men from 760 ng/dL at baseline to 295 ng/dL (60% reduction) after 24 hours and to a maximum of 123 ng/dL (85% reduction) after 36 hours.[89]
Lower dosages and concentrations of estradiol can also significantly suppress gonadotropin secretion and testosterone levels in men and transgender women.[90][91] A retrospective study of oral estradiol monotherapy in transgender women found that dosages of 1 to 8 mg/day increased mean estradiol levels to about 50 to 150 pg/mL and suppressed mean testosterone levels to about 10 to 120 ng/dL.[92] However, there was high interindividual variability in the estradiol and testosterone levels achieved, and testosterone levels were insufficiently suppressed in many even at 8 mg/day.[92] In another study, a dosage of 1 mg/day oral micronized estradiol in healthy older men, which increased circulating estradiol levels by a relatively high amount of 6-fold (to 159 pg/mL), estrone levels by 15-fold (to 386 pg/mL), and SHBG levels by 17%, was found to suppress total testosterone levels by 27% (to 436 ng/dL) and free testosterone levels by 34% (to 11.8 ng/dL).[90][91] A pharmacodynamic study of testosterone suppression by polyestradiol phosphate in men with prostate cancer found that estradiol levels of about 135 pg/mL (500 pmol/L) would decrease testosterone levels by 50% (from 430 ng/dL to 215 ng/dL), while estradiol levels of about 410 to 545 pg/mL (1500–2000 pmol/L) would decrease testosterone levels well into the castrate range to about 6 to 12 ng/dL (0.2–0.4 nmol/L).[93]
Oral conjugated estrogens at a dosage of 7.5 mg/day has been found to suppress total testosterone levels in men to an equivalent extent as 3 mg/day oral diethylstilbestrol, which is the minimum dosage of diethylstilbestrol required to consistently suppress total testosterone levels into the castrate range (<50 ng/dL).[94] The equivalent dosage in the case of oral estradiol has not been reported. However, on the basis of the results of one study, it appears to be greater than 8 mg/day.[92] In addition, oral estradiol is known to have similar or slightly lower antigonadotropic potency than oral conjugated estrogens; the potencies of oral conjugated estrogens in terms of suppression of LH and FSH levels are 1.0 and 1.1–1.3 relative to oral estradiol, respectively.[1][95]
In addition to their antigonadotropic effects, high doses of estrogens appear to have direct toxic effects in the testes.[96][97][98][99][100][101][102][103] Following long-term therapy (>3 years) with high-dose estrogen therapy, testosterone levels fail to return to normal upon discontinuation of treatment in men with prostate cancer.[96][97][98][99][100][101][102][103] Long-lasting suppression of pituitary gland function, persisting after estrogen discontinuation, may also be involved.[103] With shorter-term estrogen therapy, testicular morphology has been reported to return to normal within 18 to 24 months following estrogen discontinuation.[104]
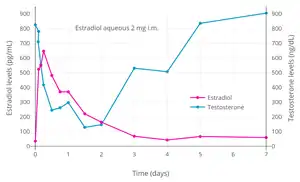
 Testosterone levels in relation to estradiol levels (and corresponding estradiol dosages) during therapy with oral estradiol alone or in combination with an antiandrogen in transgender women.[92] Dashed purple line is upper limit for female/castrate range (~50 ng/dL) and dashed grey line is testosterone level in a comparison group of post-operative transgender women (21.7 pg/mL).[92]
Testosterone levels in relation to estradiol levels (and corresponding estradiol dosages) during therapy with oral estradiol alone or in combination with an antiandrogen in transgender women.[92] Dashed purple line is upper limit for female/castrate range (~50 ng/dL) and dashed grey line is testosterone level in a comparison group of post-operative transgender women (21.7 pg/mL).[92] Estradiol and testosterone levels over the course of 12 weeks following a single intramuscular injection of 320 mg polyestradiol phosphate in men with prostate cancer.[81]
Estradiol and testosterone levels over the course of 12 weeks following a single intramuscular injection of 320 mg polyestradiol phosphate in men with prostate cancer.[81]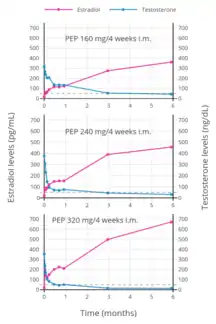 Estradiol and testosterone levels with polyestradiol phosphate 160, 240, or 320 mg once every 4 weeks by intramuscular injection in men with prostate cancer.[105]
Estradiol and testosterone levels with polyestradiol phosphate 160, 240, or 320 mg once every 4 weeks by intramuscular injection in men with prostate cancer.[105]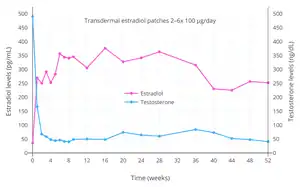
 Estradiol, testosterone, and prolactin levels with 100 mg/month estradiol undecylate by intramuscular injection in men with prostate cancer.[108]
Estradiol, testosterone, and prolactin levels with 100 mg/month estradiol undecylate by intramuscular injection in men with prostate cancer.[108]
Progonadotropic effects
Generally, estrogens are antigonadotropic and inhibit gonadotropin secretion.[109][110] However, in women, a sharp increase in estradiol levels to about 200 to 500 pg/mL occurs at the end of the follicular phase (mid-cycle) during the normal menstrual cycle and paradoxically triggers a surge in LH and FSH secretion.[109][111][110] This occurs when estradiol concentrations reach levels of about 250 to 300 pg/mL.[112] During the mid-cycle surge, LH levels increase by 3- to 12-fold and FSH levels increase by 2- to 4-fold.[113][114][115] The surge lasts about 24 to 36 hours and triggers ovulation, the rupture of the dominant ovarian follicle and the release of the egg from the ovary into the oviduct.[109] This estrogen-mediated gonadotropin surge effect has also been found to occur with exogenous estrogen, including in transgender women on hormone therapy and pre-hormone therapy transgender men acutely challenged with a high dose of an estrogen, but does not occur in men, pre-hormone therapy transgender women, or transgender men on hormone therapy, hence indicating a hormonally-based sex difference.[116] Progestogens have antiestrogenic actions on the progonadotropic effects of estrogens[117] and a sufficient amount of progesterone (corresponding to levels greater than 2 ng/mL) or a progestin prevents the mid-cycle estradiol-induced surge in gonadotropin levels in women.[118][119] This is how progestins prevent ovulation and in part mediate their contraceptive effects in women.[119]
Effects on adrenal androgen levels
In addition to their antigonadotropic effects, estrogens at high concentrations can significantly decrease androgen production by the adrenal glands.[3][120][121] A study found that treatment with a high dosage of ethinylestradiol (100 µg/day) reduced circulating adrenal androgen levels by 27 to 48% in transgender women.[3][120][121] Another study found similar effects in men with prostate cancer, with levels of the adrenal androgens dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), and androstenedione (A4) all decreasing significantly more with high-dose estrogen therapy (oral ethinylestradiol plus intramuscular polyestradiol phosphate) than with orchiectomy (by 33–39% and 10–26%, respectively).[122]
However, studies have found that these effects occur with high-dose oral and synthetic estrogens such as ethinylestradiol and estramustine phosphate but minimally with the parenteral bioidentical estrogens polyestradiol phosphate and estradiol undecylate, suggesting that decreases in adrenal androgen levels are secondary to changes in liver protein synthesis rather than due to a direct action in the adrenal cortex, and that such changes will only occur in the context of strong hepatic impact.[122][97][123] Cortisol levels were unchanged in the other groups (e.g., orchiectomy, GnRH agonist therapy, and parenteral estrogen therapy) in this study, but increased by 300 to 400% in the oral and synthetic estrogen groups, likely secondary to increases in hepatic corticosteroid-binding globulin (CBG) production and compensatory upregulation of adrenal corticosteroid synthesis.[123]
Changes in levels of weak adrenal androgens are of relevance as these androgens serve as circulating reservoir of precursors that are transformed in tissues into potent androgens like testosterone and dihydrotestosterone and into estrogens.[124][125][126][127]
Effects on liver protein synthesis
Estradiol and other estrogens modulate liver protein synthesis via activation of hepatic ERs.[1] Estradiol increases the production and by extension circulating levels of sex hormone-binding globulin (SHBG), corticosteroid-binding globulin (CBG), angiotensinogen (AGT), pregnancy zone protein (PZP), coagulation factors, and numerous other hepatic proteins.[1] Conversely, estradiol decreases hepatic synthesis and by extension circulating levels of insulin-like growth factor 1 (IGF-1).[1] The effects of estradiol on liver protein synthesis are moderated by route of administration, with oral administration having 4- or 5-fold stronger effects on liver protein synthesis than doses by the transdermal route with equivalent general/systemic estrogenic potency.[1] The influences of estradiol on liver protein synthesis have a variety of effects in the body, with implications for the bioavailability of androgens and the cardiovascular system.[1]
The influence of 2 mg/day oral estradiol on levels of hepatic proteins such as SHBG, CBG, and AGT is much lower than that with 10 μg/day oral ethinylestradiol.[128][68] Vaginal micronized estradiol at 0.25 mg/day increased SHBG levels by about 10% after 2 weeks of therapy in women.[129][130] Estradiol-containing birth control pills, which contain 1 to 3 mg/day estradiol or estradiol valerate, have been found to increase SHBG levels by 1.5-fold.[131][132] Both oral estradiol valerate at 6 mg/day and intramuscular estradiol valerate at 10 mg every 10 days have been found to increase SHBG levels by 2.5- to 3-fold in transgender women.[133][134][135] For comparison, combined birth control pills containing ethinylestradiol and a progestin with minimal androgenic or antiandrogenic activity have been found to increase SHBG levels by about 3- to 4-fold.[136] High-dose polyestradiol phosphate by intramuscular injection has been found to increase SHBG levels by about 1.5-fold.[4][68]
Estradiol valerate in oil solution by intramuscular injection has been studied in the treatment of prostate cancer.[137][138][139][140] Although parenteral estradiol has diminished effects on liver protein synthesis and by extension coagulation and cardiovascular risk compared to oral estradiol and non-bioidentical estrogens, a property attributable to its absence of disproportionate effects on the liver, sufficient doses of parenteral estradiol can nonetheless result in high estradiol concentrations in the liver and may increase coagulation and cardiovascular risk similarly.[137][138][140] Estradiol valerate at a dose of 10 to 40 mg by intramuscular injection once every 2 weeks in men with prostate cancer has been found to increase markers of coagulation and plasminogen system activation such as levels of thrombin–antithrombin complex and quantitative D-dimers.[137][138][140] Administration of daily prophylactic anticoagulation in the form of low molecular-weight heparin was able to successfully return these hemostasis markers to baseline.[137][140] Doses of estradiol valerate of 10 to 40 mg by intramuscular injection have also been used to limit bleeding in women with hemorrhage due to dysfunctional uterine bleeding, although this is due primarily to stimulation of uterine growth.[141]: 318 [142]: 60
| Proteins, general | Coagulation factors | ||
|---|---|---|---|
| Compound | Effect | Compound | Effect |
| α1-Antitrypsin | + | Antithrombin III | − |
| Albumin | − | C-reactive protein | + |
| Alkaline phosphatase | + | Coagulation factor II | + |
| Angiotensinogen | + | Coagulation factor VII | + |
| Bilirubin | + | Coagulation factor VIII | + |
| Ceruloplasmin | + | Coagulation factor IX | + |
| Corticosteroid-binding globulin (transcortin) | + | Coagulation factor X | + |
| χ-Glutamyl transpeptidase | + | Coagulation factor XII | + |
| Growth hormone | + | Fibrinogen | + |
| Growth hormone-binding protein | + | Plasminogen | + |
| Insulin-like growth factor 1 | − | Protein C | + |
| Haptoglobin | − | Prothrombin time | − |
| Leucyl aminopeptidase | + | Lipids | |
| α2-Microglobulin | + | Compound | Effect |
| Orosomucoid (α1-acid glycoprotein) | − | Apolipoprotein A | + |
| Pregnancy zone protein | + | High-density lipoprotein | + |
| Retinol-binding protein | + | Low-density lipoprotein | − |
| Sex hormone-binding globulin | + | Lecithin | + |
| Thyroxine-binding globulin | + | Total lipids | + |
| Transferrin | + | Triglycerides | + |
| Key: + = Increased. − = Decreased. Sources: See template. | |||
Other effects
Estrogens have been reported to downregulate androgen receptor expression in adipose tissue, and may thereby inhibit the effects of androgens on fat distribution.[143][144][145]
Differences from other estrogens

Estradiol has relatively low oral bioavailability of about 5%.[1] In addition, there is considerable interindividual variability in levels of estradiol achieved with oral estradiol.[1] In contrast to estradiol, the synthetic estrogen ethinylestradiol has about 45% oral bioavailability, around 80- to 200-fold greater systemic oral estrogenic potency, roughly 500- to 1,500-fold greater hepatic oral estrogenic potency, and less interindividual variability in circulating estrogen levels achieved.[68][1][147][148][149][150][151] An oral dose of ethinylestradiol that is approximately 100-fold lower than that of estradiol achieves similar maximal circulating estrogen concentrations (e.g., 50 pg/mL ethinylestradiol with a single 20 μg dose of ethinylestradiol relative to 40 pg/mL estradiol with a single 2 mg dose of micronized estradiol or estradiol valerate).[1] These differences are due to the introduction of an ethynyl group at the C17α position in ethinylestradiol (also known as 17α-ethynylestradiol), which results in steric hindrance and greatly diminishes the first-pass metabolism of ethinylestradiol relative to estradiol with oral administration.[1] Estradiol and ethinylestradiol have similar affinities for and efficacies as agonists of the ERs,[1][2] and the systemic estrogenic potency of estradiol and ethinylestradiol is similar when they are administered by the intravenous route.[152]
Synthetic estrogens like ethinylestradiol and diethylstilbestrol and the natural but animal-derived conjugated estrogens have disproportionate effects on liver protein synthesis relative to their effects in other tissues when compared to estradiol.[1] At doses via the oral route with comparable systemic estrogenic potency, conjugated estrogens have about 1.3 to 4.5 times the hepatotropic potency (i.e., potency in modulating liver protein synthesis) of estradiol, ethinylestradiol has about 2.9 to 5.0 times the hepatotropic potency of estradiol, and diethylstilbestrol shows about 5.7 to 7.5 times the hepatotropic potency of estradiol (all measured via a small selection of estrogen-modulated hepatic proteins that included HDL cholesterol, SHBG, CBG, and angiotensinogen).[1] The greater hepatotropic potency of these estrogens relative to estradiol is related to susceptibility to hepatic metabolism.[1] Whereas estradiol is metabolized and thereby inactivated rapidly upon entry into the liver, other estrogens like ethinylestradiol and diethylstilbestrol are resistant to hepatic metabolism and persist in the liver for a longer amount of time.[1] This is reflected in the biological half-lives of these estrogens; the blood half-life of estradiol is about 1 to 2 hours, while the half-lives of ethinylestradiol and diethylstilbestrol are approximately 20 hours and 24 hours, respectively.[153][154][151] In accordance with its long half-life, ethinylestradiol passes through the liver many times prior to its elimination.[155] Because humans are not adapted to efficiently metabolize conjugated estrogens (which are equine (horse) estrogens) and synthetic estrogens like ethinylestradiol and diethylstilbestrol, these estrogens are not properly inactivated in the liver, with markedly disproportionate hepatic estrogenic effects resulting.[1]
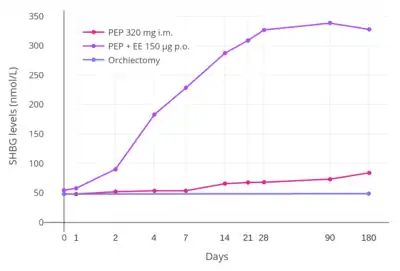
In addition to differences in hepatotropic potency between estradiol and other estrogens, there are differences in hepatotropic potency between different routes of administration of estradiol.[1] Due to the first pass through the liver, oral estradiol results in disproportionate and unphysiological hepatic estradiol levels that are 4- to 5-fold higher than in the circulation.[156][1] Conversely, parenteral routes of estradiol, such as transdermal, vaginal, and injection, bypass the first pass through the liver and produce levels of estradiol in the circulation and liver that are comparable.[156][1] As an example of the reduced hepatic impact of parenteral estradiol relative to oral estradiol, a study found that 1 mg/day oral estradiol significantly increased SHBG levels by 45%, while 50 µg/day transdermal estradiol increased SHBG levels non-significantly by only 12% (with these dosages being roughly equivalent in systemic estrogenic potency).[157][158][159] As such, not only do oral non-bioidentical estrogens like ethinylestradiol and conjugated estrogens have substantially greater potency in the liver than does oral estradiol, oral estradiol has considerably greater potency in the liver than does parenteral estradiol.[1] Thus, the hepatotropic effects of oral non-bioidentical estrogens like ethinylestradiol are massive in comparison to parenteral estradiol (see the graph above/to the right), which in contrast to these estrogens has very weak or even absent effects on liver protein synthesis at normal therapeutic dosages.[1][81][68][3] Whereas high-dosage 320 mg/month intramuscular polyestradiol phosphate increased SHBG levels to 166% in men with prostate cancer, the combination of 80 mg/month intramuscular polyestradiol phosphate and high-dosage 150 µg/day oral ethinylestradiol increased levels of SHBG to 617%, an almost 8-fold difference in increase and almost 4-fold difference in absolute levels between the two treatment regimens.[4][81][160]
The effects of estrogens on liver protein synthesis, such as on the synthesis of coagulation factors, lipoproteins, and triglycerides, can cause an increased risk of thromboembolic and cardiovascular complications, which in turn can result in increased mortality.[68] The risk of thromboembolic and cardiovascular complications is significantly increased in postmenopausal women taking oral conjugated estrogens as a component of menopausal hormone therapy.[1][161][162] Both oral estradiol and oral esterified estrogens have been found to have a significantly lower risk of thromboembolic and cardiovascular complications than oral conjugated estrogens, and transdermal estradiol appears to have no such risks at all.[1][163][161][162] Widely employed in the past, oral synthetic estrogens like ethinylestradiol and diethylstilbestrol are no longer used in menopausal hormone therapy due to their high risks of thromboembolic and cardiovascular complications.[164] Studies have found a markedly increased 5-year risk of cardiovascular mortality of 14 to 26% in men treated with high-dosage oral synthetic estrogens like ethinylestradiol and diethylstilbestrol for prostate cancer.[68] With diethylstilbestrol, there is an up to 35% incidence of cardiovascular toxicity and an up to 15% incidence of venous thromboembolism.[165] In a small study comparing high-dosage 320 mg/month intramuscular polyestradiol phosphate versus the combination of 80 mg/month polyestradiol phosphate with high-dosage 150 µg/day oral ethinylestradiol for prostate cancer, there was a 25% incidence of cardiovascular complications over the course of a year in the group that was also treated with ethinylestradiol, whereas there were no cardiovascular complications in the polyestradiol phosphate-only group.[81] In accordance, another study found no change in levels of coagulation factor VII, a protein of particular importance in the cardiovascular side effects of estrogens, with 240 mg/month intramuscular polyestradiol phosphate.[166] In spite of the markedly reduced impact of parenteral estradiol on the liver compared to other estrogens however, high dosages of parenteral estradiol, producing high levels of circulating estradiol, can still result in important and undesirable changes in liver protein synthesis as with other estrogens.[40] A high dosage of 320 mg/month polyestradiol phosphate has been found to result in significantly increased cardiovascular morbidity (due to non-fatal ischemic heart events and heart decompensation) in men with prostate cancer in two large studies, though cardiovascular mortality was notably not increased.[40][167]
In addition to the liver, ethinylestradiol shows disproportionate estrogenic effects in the uterus.[1][43][168] This is due to its inability to be inactivated by uterine 17β-hydroxysteroid dehydrogenase (17β-HSD).[1][43][168] Because of its disproportionate effects in the uterus, ethinylestradiol is associated with a significantly lower incidence of vaginal bleeding and spotting than is estradiol, particularly in combination with progestogens (which induce 17β-HSD expression and hence estradiol metabolism in the uterus),[1] and is an important contributing factor in why ethinylestradiol, among other reasons and in spite of its inferior safety profile, has been widely used in oral contraceptives instead of estradiol.[132][131] Although ethinylestradiol has increased effects in the uterus relative to estradiol, it is similarly not associated with an increase in the risk of endometrial hyperplasia and endometrial cancer when used in combination with a progestogen, but instead with a significant decrease.[1][169]
See also
References
- Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration" (PDF). Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947. S2CID 24616324.
- Michael Oettel; Ekkehard Schillinger (6 December 2012). Estrogens and Antiestrogens I: Physiology and Mechanisms of Action of Estrogens and Antiestrogens. Springer Science & Business Media. pp. 121, 226, 235–237. ISBN 978-3-642-58616-3.
- Michael Oettel; Ekkehard Schillinger (6 December 2012). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen. Springer Science & Business Media. pp. 163–178, 235–237, 252–253, 261–276, 538–543. ISBN 978-3-642-60107-1.
- Stege R, Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A (1988). "Single drug polyestradiol phosphate therapy in prostatic cancer". Am. J. Clin. Oncol. 11 Suppl 2: S101–3. doi:10.1097/00000421-198801102-00024. PMID 3242384. S2CID 32650111.
- Ockrim JL, Lalani EN, Laniado ME, Carter SS, Abel PD (2003). "Transdermal estradiol therapy for advanced prostate cancer--forward to the past?". J. Urol. 169 (5): 1735–7. doi:10.1097/01.ju.0000061024.75334.40. PMID 12686820.
- Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (2006). "Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta". Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID 16554039.
- Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S (July 1998). "Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists". Mol. Pharmacol. 54 (1): 105–12. doi:10.1124/mol.54.1.105. PMID 9658195.
- Prossnitz ER, Arterburn JB (July 2015). "International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators". Pharmacol. Rev. 67 (3): 505–40. doi:10.1124/pr.114.009712. PMC 4485017. PMID 26023144.
- Soltysik K, Czekaj P (April 2013). "Membrane estrogen receptors – is it an alternative way of estrogen action?". J. Physiol. Pharmacol. 64 (2): 129–42. PMID 23756388.
- Prossnitz ER, Barton M (May 2014). "Estrogen biology: New insights into GPER function and clinical opportunities". Mol. Cell. Endocrinol. 389 (1–2): 71–83. doi:10.1016/j.mce.2014.02.002. PMC 4040308. PMID 24530924.
- A. Labhart (6 December 2012). Clinical Endocrinology: Theory and Practice. Springer Science & Business Media. pp. 548, 551. ISBN 978-3-642-96158-8.
- Ojasoo T, Raynaud JP (November 1978). "Unique steroid congeners for receptor studies". Cancer Res. 38 (11 Pt 2): 4186–98. PMID 359134.
- Ojasoo T, Delettré J, Mornon JP, Turpin-VanDycke C, Raynaud JP (1987). "Towards the mapping of the progesterone and androgen receptors". J. Steroid Biochem. 27 (1–3): 255–69. doi:10.1016/0022-4731(87)90317-7. PMID 3695484.
- Raynaud JP, Bouton MM, Moguilewsky M, Ojasoo T, Philibert D, Beck G, Labrie F, Mornon JP (January 1980). "Steroid hormone receptors and pharmacology". J. Steroid Biochem. 12: 143–57. doi:10.1016/0022-4731(80)90264-2. PMID 7421203.
- Raynaud, J.P.; Ojasoo, T.; Bouton, M.M.; Philibert, D. (1979). Drug Design. pp. 169–214. doi:10.1016/B978-0-12-060308-4.50010-X. ISBN 9780120603084.
- Blankvoort BM, de Groene EM, van Meeteren-Kreikamp AP, Witkamp RF, Rodenburg RJ, Aarts JM (November 2001). "Development of an androgen reporter gene assay (AR-LUX) utilizing a human cell line with an endogenously regulated androgen receptor". Anal. Biochem. 298 (1): 93–102. doi:10.1006/abio.2001.5352. PMID 11673900.
- Eberhard Nieschlag; Hermann M. Behre; Susan Nieschlag (26 July 2012). Testosterone: Action, Deficiency, Substitution. Cambridge University Press. pp. 495–. ISBN 978-1-107-01290-5.
- Goldstein I, Meston CM, Davis S, Traish A (17 November 2005). Women's Sexual Function and Dysfunction: Study, Diagnosis and Treatment. CRC Press. pp. 205–, 540. ISBN 978-1-84214-263-9.
- Robert Marcus; David W. Dempster; Jane A. Cauley; David Feldman (13 June 2013). Osteoporosis. Academic Press. pp. 1117–. ISBN 978-0-12-398252-0.
Altogether, men make 20-fold more androgens than do women; the proportion of androgen converted to E2 is 200-fold more in women; and E2 is 1000-fold more potent than androgens (on a molar basis) on target tissues [28]. Thus, circulating estrogen levels are measured in picograms, and testosterone levels are measured in nanograms.
- Thomas, John A.; Keenan, Edward J. (1986). Principles of Endocrine Pharmacology. pp. 135–165. doi:10.1007/978-1-4684-5036-1_7. ISBN 978-0-306-42143-3.
Cytoplasmic estrogen receptors characteristically exhibit high affinity for estradiol-17J3, with an equilibrium dissociation constant of 0.1 nM. The number of these sites in target tissues is generally low, approximating 10,000-20,000 sites per cell.
- Wibowo E, Schellhammer P, Wassersug RJ (January 2011). "Role of estrogen in normal male function: clinical implications for patients with prostate cancer on androgen deprivation therapy". J. Urol. 185 (1): 17–23. doi:10.1016/j.juro.2010.08.094. PMID 21074215.
In cell culture37 and gonadectomized rodents48 the addition of E can induce the autoregulation of ERs. This finding suggests that the ER expression depends on the level of serum E and to maintain an effective cellular response to E2 regulation of the ER is crucial. Prolonged E2 administration at a constant dose may not be maximally effective for patients with PCa. As a result of continuous exposure, ERs may be down-regulated, attenuating their effectiveness. Thus, cyclical rather than continuous administration of E may be preferable.
- Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW (March 1999). "Proteasome-dependent degradation of the human estrogen receptor". Proc. Natl. Acad. Sci. U.S.A. 96 (5): 1858–62. Bibcode:1999PNAS...96.1858N. doi:10.1073/pnas.96.5.1858. PMC 26701. PMID 10051559.
- Miller, Colette (October 2015). "A brief on the structure and function of estrogen receptor alpha (BCMB8010 Enzyme Project)". doi:10.13140/RG.2.1.4082.5044.
ERα is relatively stable in the cell with a half-life of up to 5 days, however once bound to ligand this time shortens to 3-4 hours.
{{cite journal}}: Cite journal requires|journal=(help) - Kloosterboer, Helenius; Schoonen, Willem; Verheul, Herman (2008). Breast Cancer. The Oncologist. Vol. 17. pp. 343–366. doi:10.3109/9781420058734-19. ISBN 978-1-4200-5872-7. PMC 3267821. PMID 22234628.
Steroid deprivation, for instance, can have a major impact on the growth stimulation by E2. Estrogen sensitivity can be increased easily by four log-units or more (Masamura et al., 1995; Chan et al., 2002) (Fig. 1). This effect may be explained, at least partly, by a 100-fold higher level of ER(s) (Zajchowski et al., 1993), but coactivator sensitivity as well as the degree of phosphorylation of transactivation factors (TAF-1 and/or TAF-2) may also be crucial.
- Mauvais-Jarvis P, Kuttenn F, Gompel A (1986). "Antiestrogen action of progesterone in breast tissue". Breast Cancer Res. Treat. 8 (3): 179–88. doi:10.1007/BF01807330. PMID 3297211. S2CID 17511105.
- Zhou J, Ng S, Adesanya-Famuiya O, Anderson K, Bondy CA (September 2000). "Testosterone inhibits estrogen-induced mammary epithelial proliferation and suppresses estrogen receptor expression". FASEB J. 14 (12): 1725–30. doi:10.1096/fj.99-0863com. PMID 10973921. S2CID 17172449.
- Weigel, Nancy L.; Smith, Carolyn L. (2016). "Estrogen and Progesterone Action". Endocrinology: Adult and Pediatric. pp. 2207–2215.e3. doi:10.1016/B978-0-323-18907-1.00127-X. ISBN 9780323189071.
- Purohit A, Woo LW, Potter BV (July 2011). "Steroid sulfatase: a pivotal player in estrogen synthesis and metabolism" (PDF). Mol. Cell. Endocrinol. 340 (2): 154–60. doi:10.1016/j.mce.2011.06.012. PMID 21693170. S2CID 14296237.
- Africander D, Storbeck KH (May 2018). "Steroid metabolism in breast cancer: Where are we and what are we missing?". Mol. Cell. Endocrinol. 466: 86–97. doi:10.1016/j.mce.2017.05.016. PMID 28527781. S2CID 4547808.
- Mueller JW, Gilligan LC, Idkowiak J, Arlt W, Foster PA (October 2015). "The Regulation of Steroid Action by Sulfation and Desulfation". Endocr. Rev. 36 (5): 526–63. doi:10.1210/er.2015-1036. PMC 4591525. PMID 26213785.
- Klinge CM (July 2001). "Estrogen receptor interaction with estrogen response elements". Nucleic Acids Res. 29 (14): 2905–19. doi:10.1093/nar/29.14.2905. PMC 55815. PMID 11452016.
- Christian Behl (22 June 2001). Estrogen — Mystery Drug for the Brain?: The Neuroprotective Activities of the Female Sex Hormone. Springer Science & Business Media. pp. 41–. ISBN 978-3-211-83539-5.
- Fritz F. Parl (2000). Estrogens, Estrogen Receptor and Breast Cancer. IOS Press. pp. 4, 111. ISBN 978-0-9673355-4-4.
- Jennifer E. Dietrich (18 June 2014). Female Puberty: A Comprehensive Guide for Clinicians. Springer. pp. 53–. ISBN 978-1-4939-0912-4.
- Randy Thornhill; Steven W. Gangestad (25 September 2008). The Evolutionary Biology of Human Female Sexuality. Oxford University Press. pp. 145–. ISBN 978-0-19-988770-5.
- Raine-Fenning NJ, Brincat MP, Muscat-Baron Y (2003). "Skin aging and menopause : implications for treatment". Am J Clin Dermatol. 4 (6): 371–8. doi:10.2165/00128071-200304060-00001. PMID 12762829. S2CID 20392538.
- Chris Hayward (31 July 2003). Gender Differences at Puberty. Cambridge University Press. pp. 22–. ISBN 978-0-521-00165-6.
- Shlomo Melmed; Kenneth S. Polonsky; P. Reed Larsen; Henry M. Kronenberg (11 November 2015). Williams Textbook of Endocrinology. Elsevier Health Sciences. pp. 1105–. ISBN 978-0-323-34157-8.
- Richard E. Jones; Kristin H. Lopez (28 September 2013). Human Reproductive Biology. Academic Press. pp. 19–. ISBN 978-0-12-382185-0.
- Waun Ki Hong; James F. Holland (2010). Holland-Frei Cancer Medicine 8. PMPH-USA. pp. 753–. ISBN 978-1-60795-014-1.
- Ethel Sloane (2002). Biology of Women. Cengage Learning. pp. 496–. ISBN 978-0-7668-1142-3.
- Tekoa L. King; Mary C. Brucker (25 October 2010). Pharmacology for Women's Health. Jones & Bartlett Learning. pp. 1022–. ISBN 978-0-7637-5329-0.
- Rogerio A. Lobo (5 June 2007). Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. Academic Press. pp. 177, 217–226, 770–771. ISBN 978-0-08-055309-2.
- David Warshawsky; Joseph R. Landolph Jr. (31 October 2005). Molecular Carcinogenesis and the Molecular Biology of Human Cancer. CRC Press. pp. 457–. ISBN 978-0-203-50343-0.
- Acevedo-Rodriguez A, Mani SK, Handa RJ (2015). "Oxytocin and Estrogen Receptor β in the Brain: An Overview". Frontiers in Endocrinology. 6: 160. doi:10.3389/fendo.2015.00160. PMC 4606117. PMID 26528239.
- Lauritzen C (September 1990). "Clinical use of oestrogens and progestogens". Maturitas. 12 (3): 199–214. doi:10.1016/0378-5122(90)90004-P. PMID 2215269.
- Lauritzen C (June 1977). "[Estrogen thearpy in practice. 3. Estrogen preparations and combination preparations]" [Estrogen therapy in practice. 3. Estrogen preparations and combination preparations]. Fortschritte Der Medizin (in German). 95 (21): 1388–92. PMID 559617.
- Wolf AS, Schneider HP (12 March 2013). Östrogene in Diagnostik und Therapie. Springer-Verlag. pp. 78–. ISBN 978-3-642-75101-1.
- Göretzlehner G, Lauritzen C, Römer T, Rossmanith W (1 January 2012). Praktische Hormontherapie in der Gynäkologie. Walter de Gruyter. pp. 44–. ISBN 978-3-11-024568-4.
- Knörr K, Beller FK, Lauritzen C (17 April 2013). Lehrbuch der Gynäkologie. Springer-Verlag. pp. 212–213. ISBN 978-3-662-00942-0.
- Horský J, Presl J (1981). "Hormonal Treatment of Disorders of the Menstrual Cycle". In Horsky J, Presl J (eds.). Ovarian Function and its Disorders: Diagnosis and Therapy. Springer Science & Business Media. pp. 309–332. doi:10.1007/978-94-009-8195-9_11. ISBN 978-94-009-8195-9.
- Pschyrembel W (1968). Praktische Gynäkologie: für Studierende und Ärzte. Walter de Gruyter. pp. 598–599. ISBN 978-3-11-150424-7.
- Lauritzen CH (January 1976). "The female climacteric syndrome: significance, problems, treatment". Acta Obstetricia Et Gynecologica Scandinavica. Supplement. 51: 47–61. doi:10.3109/00016347509156433. PMID 779393.
- Lauritzen C (1975). "The Female Climacteric Syndrome: Significance, Problems, Treatment". Acta Obstetricia et Gynecologica Scandinavica. 54 (s51): 48–61. doi:10.3109/00016347509156433. ISSN 0001-6349.
- Kopera H (1991). "Hormone der Gonaden". Hormonelle Therapie für die Frau. Kliniktaschenbücher. pp. 59–124. doi:10.1007/978-3-642-95670-6_6. ISBN 978-3-540-54554-5. ISSN 0172-777X.
- Scott WW, Menon M, Walsh PC (April 1980). "Hormonal Therapy of Prostatic Cancer". Cancer. 45 Suppl 7: 1929–1936. doi:10.1002/cncr.1980.45.s7.1929. PMID 29603164.
- Leinung MC, Feustel PJ, Joseph J (2018). "Hormonal Treatment of Transgender Women with Oral Estradiol". Transgender Health. 3 (1): 74–81. doi:10.1089/trgh.2017.0035. PMC 5944393. PMID 29756046.
- Ryden AB (1950). "Natural and synthetic oestrogenic substances; their relative effectiveness when administered orally". Acta Endocrinologica. 4 (2): 121–39. doi:10.1530/acta.0.0040121. PMID 15432047.
- Ryden AB (1951). "The effectiveness of natural and synthetic oestrogenic substances in women". Acta Endocrinologica. 8 (2): 175–91. doi:10.1530/acta.0.0080175. PMID 14902290.
- Kottmeier HL (1947). "Ueber blutungen in der menopause: Speziell der klinischen bedeutung eines endometriums mit zeichen hormonaler beeinflussung: Part I". Acta Obstetricia et Gynecologica Scandinavica. 27 (s6): 1–121. doi:10.3109/00016344709154486. ISSN 0001-6349.
There is no doubt that the conversion of the endometrium with injections of both synthetic and native estrogenic hormone preparations succeeds, but the opinion whether native, orally administered preparations can produce a proliferation mucosa changes with different authors. PEDERSEN-BJERGAARD (1939) was able to show that 90% of the folliculin taken up in the blood of the vena portae is inactivated in the liver. Neither KAUFMANN (1933, 1935), RAUSCHER (1939, 1942) nor HERRNBERGER (1941) succeeded in bringing a castration endometrium into proliferation using large doses of orally administered preparations of estrone or estradiol. Other results are reported by NEUSTAEDTER (1939), LAUTERWEIN (1940) and FERIN (1941); they succeeded in converting an atrophic castration endometrium into an unambiguous proliferation mucosa with 120–300 oestradiol or with 380 oestrone.
- Rietbrock N, Staib AH, Loew D (11 March 2013). Klinische Pharmakologie: Arzneitherapie. Springer-Verlag. pp. 426–. ISBN 978-3-642-57636-2.
- Martinez-Manautou J, Rudel HW (1966). "Antiovulatory Activity of Several Synthetic and Natural Estrogens". In Robert Benjamin Greenblatt (ed.). Ovulation: Stimulation, Suppression, and Detection. Lippincott. pp. 243–253.
- Herr, F.; Revesz, C.; Manson, A. J.; Jewell, J. B. (1970). "Biological Properties of Estrogen Sulfates". Chemical and Biological Aspects of Steroid Conjugation. pp. 368–408. doi:10.1007/978-3-642-49793-3_8. ISBN 978-3-642-49506-9.
- Duncan CJ, Kistner RW, Mansell H (October 1956). "Suppression of ovulation by trip-anisyl chloroethylene (TACE)". Obstetrics and Gynecology. 8 (4): 399–407. PMID 13370006.
- Scherr DS, Pitts WR (2003). "The nonsteroidal effects of diethylstilbestrol: the rationale for androgen deprivation therapy without estrogen deprivation in the treatment of prostate cancer". J. Urol. 170 (5): 1703–8. doi:10.1097/01.ju.0000077558.48257.3d. PMID 14532759.
- Coss, Christopher C.; Jones, Amanda; Parke, Deanna N.; Narayanan, Ramesh; Barrett, Christina M.; Kearbey, Jeffrey D.; Veverka, Karen A.; Miller, Duane D.; Morton, Ronald A.; Steiner, Mitchell S.; Dalton, James T. (2012). "Preclinical Characterization of a Novel Diphenyl Benzamide Selective ERα Agonist for Hormone Therapy in Prostate Cancer". Endocrinology. 153 (3): 1070–1081. doi:10.1210/en.2011-1608. ISSN 0013-7227. PMID 22294742.
- Novara G, Galfano A, Secco S, Ficarra V, Artibani W (2009). "Impact of surgical and medical castration on serum testosterone level in prostate cancer patients". Urol. Int. 82 (3): 249–55. doi:10.1159/000209352. PMID 19440008. S2CID 24771328.
- von Schoultz B, Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A, Stege R (1989). "Estrogen therapy and liver function--metabolic effects of oral and parenteral administration". Prostate. 14 (4): 389–95. doi:10.1002/pros.2990140410. PMID 2664738. S2CID 21510744.
- Wein AJ, Kavoussi LR, Novick AC, Partin AW, Peters CA (25 August 2011). Campbell-Walsh Urology: Expert Consult Premium Edition: Enhanced Online Features and Print, 4-Volume Set. Elsevier Health Sciences. pp. 2938–. ISBN 978-1-4160-6911-9.
- Knuth UA, Hano R, Nieschlag E (1984). "Effect of flutamide or cyproterone acetate on pituitary and testicular hormones in normal men". J. Clin. Endocrinol. Metab. 59 (5): 963–9. doi:10.1210/jcem-59-5-963. PMID 6237116.
- Jacobi GH, Altwein JE, Kurth KH, Basting R, Hohenfellner R (1980). "Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial". Br J Urol. 52 (3): 208–15. doi:10.1111/j.1464-410x.1980.tb02961.x. PMID 7000222.
- Sander S, Nissen-Meyer R, Aakvaag A (1978). "On gestagen treatment of advanced prostatic carcinoma". Scand. J. Urol. Nephrol. 12 (2): 119–21. doi:10.3109/00365597809179977. PMID 694436.
- Kjeld JM, Puah CM, Kaufman B, Loizou S, Vlotides J, Gwee HM, Kahn F, Sood R, Joplin GF (1979). "Effects of norgestrel and ethinyloestradiol ingestion on serum levels of sex hormones and gonadotrophins in men". Clin. Endocrinol. (Oxf). 11 (5): 497–504. doi:10.1111/j.1365-2265.1979.tb03102.x. PMID 519881. S2CID 5836155.
- Watson NR, Studd JW, Riddle AF, Savvas M (October 1988). "Suppression of ovulation by transdermal oestradiol patches". BMJ. 297 (6653): 900–1. doi:10.1136/bmj.297.6653.900. PMC 1834440. PMID 3140971.
- Sitruk-Ware R (June 1995). "Transdermal application of steroid hormones for contraception". J. Steroid Biochem. Mol. Biol. 53 (1–6): 247–51. doi:10.1016/0960-0760(95)00055-5. PMID 7626463. S2CID 30461300.
- Studd, J. (2012). "Treatment of premenstrual disorders by suppression of ovulation by transdermal estrogens". Menopause International. 18 (2): 65–67. doi:10.1258/mi.2012.012015. ISSN 1754-0453. PMID 22611224. S2CID 8914354.
- Toppozada M (June 1977). "The clinical use of monthly injectable contraceptive preparations". Obstet Gynecol Surv. 32 (6): 335–47. doi:10.1097/00006254-197706000-00001. PMID 865726.
- el-Mahgoub S, Karim M (February 1972). "Depot estrogen as a monthly contraceptive in nulliparous women with mild uterine hypoplasia". Am. J. Obstet. Gynecol. 112 (4): 575–6. doi:10.1016/0002-9378(72)90319-5. PMID 5008627.
- Jorge Martinez-Manautou; Harry W. Rudel (1966). "Antiovulatory Activity of Several Synthetic and Natural Estrogens". In Robert Benjamin Greenblatt (ed.). Ovulation: Stimulation, Suppression, and Detection. Lippincott. pp. 243–253. ISBN 9780397590100.
- Herr, F.; Revesz, C.; Manson, A. J.; Jewell, J. B. (1970). "Biological Properties of Estrogen Sulfates". Chemical and Biological Aspects of Steroid Conjugation. pp. 368–408. doi:10.1007/978-3-642-49793-3_8. ISBN 978-3-642-49506-9.
- Stege R, Gunnarsson PO, Johansson CJ, Olsson P, Pousette A, Carlström K (May 1996). "Pharmacokinetics and testosterone suppression of a single dose of polyestradiol phosphate (Estradurin) in prostatic cancer patients". Prostate. 28 (5): 307–10. doi:10.1002/(SICI)1097-0045(199605)28:5<307::AID-PROS6>3.0.CO;2-8. PMID 8610057. S2CID 33548251.
- Gokhan Ozyigit; Ugur Selek (1 August 2017). Principles and Practice of Urooncology: Radiotherapy, Surgery and Systemic Therapy. Springer. pp. 334–. ISBN 978-3-319-56114-1.
The castrate level was defined as testosterone being less than 50 ng/dL (1.7 nmol/L), many years ago. However contemporary laboratory testing methods showed that the mean value after surgical castration is 15 ng/dL [1]. Thus, recently the level is defined as being less than 20 ng/dL (1 nmol/L).
- Lycette JL, Bland LB, Garzotto M, Beer TM (2006). "Parenteral estrogens for prostate cancer: can a new route of administration overcome old toxicities?". Clin Genitourin Cancer. 5 (3): 198–205. doi:10.3816/CGC.2006.n.037. PMID 17239273.
- Altwein, J. (1983). "Controversial Aspects of Hormone Manipulation in Prostatic Carcinoma". Cancer of the Prostate and Kidney. pp. 305–316. doi:10.1007/978-1-4684-4349-3_38. ISBN 978-1-4684-4351-6.
- Ockrim JL; Lalani el-N; Kakkar AK; Abel PD (August 2005). "Transdermal estradiol therapy for prostate cancer reduces thrombophilic activation and protects against thromboembolism". J. Urol. 174 (2): 527–33, discussion 532–3. doi:10.1097/01.ju.0000165567.99142.1f. PMID 16006886.
- Moore E, Wisniewski A, Dobs A (2003). "Endocrine treatment of transsexual people: a review of treatment regimens, outcomes, and adverse effects". J. Clin. Endocrinol. Metab. 88 (8): 3467–73. doi:10.1210/jc.2002-021967. PMID 12915619.
- Tangpricha V, den Heijer M (2017). "Oestrogen and anti-androgen therapy for transgender women". Lancet Diabetes Endocrinol. 5 (4): 291–300. doi:10.1016/S2213-8587(16)30319-9. PMC 5366074. PMID 27916515.
- Deutsch MB, Bhakri V, Kubicek K (2015). "Effects of cross-sex hormone treatment on transgender women and men". Obstet Gynecol. 125 (3): 605–10. doi:10.1097/AOG.0000000000000692. PMC 4442681. PMID 25730222.
- Jones TM, Fang VS, Landau RL, Rosenfield R (December 1978). "Direct inhibition of Leydig cell function by estradiol". J. Clin. Endocrinol. Metab. 47 (6): 1368–73. doi:10.1210/jcem-47-6-1368. PMID 122429.
- Taxel P, Kennedy D, Fall P, Willard A, Shoukri K, Clive J, Raisz LG (2000). "The effect of short-term treatment with micronized estradiol on bone turnover and gonadotrophins in older men". Endocr. Res. 26 (3): 381–98. doi:10.3109/07435800009066175. PMID 11019903. S2CID 45695901.
- Dukes, M.N.G. (2002). Sex hormones and related compounds, including hormonal contraceptives. Side Effects of Drugs Annual. Vol. 25. pp. 478–502. doi:10.1016/S0378-6080(02)80047-2. ISBN 9780444506740. ISSN 0378-6080.
- Leinung MC, Feustel PJ, Joseph J (2018). "Hormonal Treatment of Transgender Women with Oral Estradiol". Transgend Health. 3 (1): 74–81. doi:10.1089/trgh.2017.0035. PMC 5944393. PMID 29756046.
- Johansson CJ, Gunnarsson PO (June 2000). "Pharmacodynamic model of testosterone suppression after intramuscular depot estrogen therapy in prostate cancer". Prostate. 44 (1): 26–30. doi:10.1002/1097-0045(20000615)44:1<26::AID-PROS4>3.0.CO;2-P. PMID 10861754. S2CID 30678644.
- Scott WW, Menon M, Walsh PC (April 1980). "Hormonal Therapy of Prostatic Cancer". Cancer. 45 Suppl 7: 1929–1936. doi:10.1002/cncr.1980.45.s7.1929. PMID 29603164. S2CID 4492779.
- Alfred S. Wolf; H.P.G. Schneider (12 March 2013). Östrogene in Diagnostik und Therapie. Springer-Verlag. pp. 78–. ISBN 978-3-642-75101-1.
- Salam MA (2003). Principles & Practice of Urology: A Comprehensive Text. Universal-Publishers. pp. 684–. ISBN 978-1-58112-412-5.
Estrogens act primarily through negative feedback at the hypothalamic-pituitary level to reduce LH secretion and testicular androgen synthesis. [...] Interestingly, if the treatment with estrogens is discontinued after 3 yr. of uninterrupted exposure, serum testosterone may remain at castration levels for up to another 3 yr. This prolonged suppression is thought to result from a direct effect of estrogens on the Leydig cells.
- Cox RL, Crawford ED (December 1995). "Estrogens in the treatment of prostate cancer". J. Urol. 154 (6): 1991–8. doi:10.1016/S0022-5347(01)66670-9. PMID 7500443.
- Tomić R, Bergman B (October 1987). "Hormonal effects of cessation of estrogen treatment for prostatic carcinoma". J. Urol. 138 (4): 801–3. doi:10.1016/S0022-5347(17)43379-9. PMID 3116281.
- Tomić R, Bergman B, Damber JE (February 1983). "Testicular endocrine function after withdrawal of oestrogen treatment in patients with carcinoma of the prostate". Br J Urol. 55 (1): 42–7. doi:10.1111/j.1464-410X.1983.tb07077.x. PMID 6402048.
- Daehlin L, Tomić R, Damber JE (1988). "Depressed testosterone release from testicular tissue in vitro after withdrawal of oestrogen treatment in patients with prostatic carcinoma". Scand. J. Urol. Nephrol. 22 (1): 11–3. doi:10.1080/00365599.1988.11690376. PMID 3387906.
- Tomić R, Damber JE, Bergman B (1988). "Endocrine effects of oestrogen withdrawal in long-term treated patients with prostatic adenocarcinoma". Eur. Urol. 14 (1): 6–8. doi:10.1159/000472886. PMID 3342807.
- Wortsman J, Hamidinia A, Winters SJ (June 1989). "Hypogonadism following long-term treatment with diethylstilbestrol". Am. J. Med. Sci. 297 (6): 365–8. doi:10.1097/00000441-198906000-00006. PMID 2500019. S2CID 22686874.
- Tomić R (October 1987). "Pituitary function after orchiectomy in patients with or without earlier estrogen treatment for prostatic carcinoma". J. Endocrinol. Invest. 10 (5): 479–82. doi:10.1007/BF03348174. PMID 3123547. S2CID 25897203.
- Janet Brotherton (1976). Sex Hormone Pharmacology. Academic Press. p. 341. ISBN 978-0-12-137250-7.
- Stege R, Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A (1988). "Single drug polyestradiol phosphate therapy in prostatic cancer". Am. J. Clin. Oncol. 11 Suppl 2: S101–3. doi:10.1097/00000421-198801102-00024. PMID 3242384. S2CID 32650111.
- Langley RE, Godsland IF, Kynaston H, Clarke NW, Rosen SD, Morgan RC, Pollock P, Kockelbergh R, Lalani EN, Dearnaley D, Parmar M, Abel PD (August 2008). "Early hormonal data from a multicentre phase II trial using transdermal oestrogen patches as first-line hormonal therapy in patients with locally advanced or metastatic prostate cancer". BJU Int. 102 (4): 442–5. doi:10.1111/j.1464-410X.2008.07583.x. PMC 2564109. PMID 18422771.
Available information suggests that for preparations delivering 100 µg/day of oestradiol transdermally (including the Progynova [TS forte] patches used in the original pilot study [5]) [...]
- Ockrim J, Lalani E, Abel P (October 2006). "Therapy Insight: parenteral estrogen treatment for prostate cancer--a new dawn for an old therapy". Nat Clin Pract Oncol. 3 (10): 552–63. doi:10.1038/ncponc0602. PMID 17019433. S2CID 6847203.
- Jacobi, G.H.; Altwein, J.E. (1979). "Bromocriptin als Palliativtherapie beim fortgeschrittenen Prostatakarzinom:Experimentelles und klinisches Profil eines Medikamentes" [Bromocriptine as Palliative Therapy in Advanced Prostate Cancer: Experimental and Clinical Profile of a Drugjournal=Urologia Internationalis]. Urologia Internationalis. 34 (4): 266–290. doi:10.1159/000280272. PMID 89747.
- McDowell, Julie (2010). Encyclopedia of Human Body Systems. ABC-CLIO. pp. 201–. ISBN 978-0-313-39175-0.
- Herbison AE (June 1998). "Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons". Endocr. Rev. 19 (3): 302–30. doi:10.1210/edrv.19.3.0332. PMID 9626556.
For the greater part of the ovarian cycle, estrogen helps restrain LH secretion through what has been termed its “negative feedback” action. This has been shown to occur, in part, through an inhibition of GnRH secretion in several species (7, 11–13), but also involves potent actions of estrogen on the pituitary gonadotrophs (3, 4, 14). Estrogen also exhibits a “positive feedback” influence upon the GnRH neurons and pituitary gonadotrophs to generate the preovulatory LH surge.
- Jerome Frank Strauss; Robert L. Barbieri (1 January 2009). Yen and Jaffe's Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management. Elsevier Health Sciences. pp. 807–. ISBN 978-1-4160-4907-4.
- Grunwald, K.; Rabe, T.; Runnebaum, B. (1997). "Physiology of the Menstrual Cycle". Manual on Assisted Reproduction. pp. 22–77. doi:10.1007/978-3-662-00763-1_3. ISBN 978-3-662-00765-5.
- Anand Kumar; Mona Sharma (24 July 2017). Basics of Human Andrology: A Textbook. Springer. pp. 395–. ISBN 978-981-10-3695-8.
- Miranda A. Farage; Howard I. Maibach (27 March 2017). The Vulva: Physiology and Clinical Management, Second Edition. CRC Press. pp. 139–. ISBN 978-1-4987-5245-9.
- Rogerio A. Lobo; David M Gershenson; Gretchen M Lentz; Fidel A Valea (22 June 2016). Comprehensive Gynecology E-Book. Elsevier Health Sciences. pp. 97–. ISBN 978-0-323-43003-6.
- Linda Garnets; Douglas Kimmel (6 May 2003). Psychological Perspectives on Lesbian, Gay, and Bisexual Experiences. Columbia University Press. pp. 62–. ISBN 978-0-231-50494-2.
- Römmler A, Baumgarten S, Schwartz U, Hammerstein J (June 1982). "Anti-estrogenic effects of contraceptive progestins on the dynamics of gonadotropin release". Contraception. 25 (6): 619–27. doi:10.1016/0010-7824(82)90063-4. PMID 6214371.
- Leon Speroff; Marc A. Fritz (2005). Clinical Gynecologic Endocrinology and Infertility. Lippincott Williams & Wilkins. pp. 211–. ISBN 978-0-7817-4795-0.
- Stefan Offermanns; W. Rosenthal (14 August 2008). Encyclopedia of Molecular Pharmacology. Springer Science & Business Media. pp. 388–. ISBN 978-3-540-38916-3.
- Andrew N. Margioris; George P. Chrousos (20 April 2001). Adrenal Disorders. Springer Science & Business Media. pp. 84–. ISBN 978-1-59259-101-5.
- Polderman KH, Gooren LJ, van der Veen EA (October 1995). "Effects of gonadal androgens and oestrogens on adrenal androgen levels". Clin. Endocrinol. (Oxf). 43 (4): 415–21. doi:10.1111/j.1365-2265.1995.tb02611.x. PMID 7586614. S2CID 6815423.
- Stege R, Eriksson A, Henriksson P, Carlström K (August 1987). "Orchidectomy or oestrogen treatment in prostatic cancer: effects on serum levels of adrenal androgens and related steroids". Int. J. Androl. 10 (4): 581–7. doi:10.1111/j.1365-2605.1987.tb00357.x. PMID 2958420.
Oestrogens at high doses (i.e. polyoestradiol phosphate + oral ethinyl oestradiol or high doses of diethyl stilboestrol phosphate) are reported to cause pronounced decreases in the circulating levels of DHA, DHAS and A-4 (Jonsson ef al., 1975; Leinonen et al., 1981). [...] With respect to oestrogen treatment, modest doses of polyoestradiol phosphate or oestradiol undecylate administered intramuscularly are reported to cause a slight decrease in A-4 and a slightly decreased, or unchanged, level of DHAS, while the levels of DHA remained unchanged (Jonsson et al., 1975; Luukkarinen et al., 1977; Leinonen ef al., 1981; Vermeulen et al., 1982a; Schurmeyer et al., 1986).
- Pousette A, Carlström K, Stege R (1989). "Androgens during different modes of endocrine treatment of prostatic cancer". Urol. Res. 17 (2): 95–8. doi:10.1007/BF00262027. PMID 2734983. S2CID 25309877.
- Labrie F, Martel C, Bélanger A, Pelletier G (April 2017). "Androgens in women are essentially made from DHEA in each peripheral tissue according to intracrinology". J. Steroid Biochem. Mol. Biol. 168: 9–18. doi:10.1016/j.jsbmb.2016.12.007. PMID 28153489. S2CID 2620899.
- Chang KH, Ercole CE, Sharifi N (September 2014). "Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences". Br. J. Cancer. 111 (7): 1249–54. doi:10.1038/bjc.2014.268. PMC 4183835. PMID 24867689.
- Labrie F, Bélanger A, Pelletier G, Martel C, Archer DF, Utian WH (June 2017). "Science of intracrinology in postmenopausal women". Menopause. 24 (6): 702–712. doi:10.1097/GME.0000000000000808. PMID 28098598. S2CID 3794402.
- Warner M, Gustafsson JA (January 2015). "DHEA - a precursor of ERβ ligands". J. Steroid Biochem. Mol. Biol. 145: 245–7. doi:10.1016/j.jsbmb.2014.08.003. PMID 25125389. S2CID 26043868.
- Mashchak CA, Lobo RA, Dozono-Takano R, Eggena P, Nakamura RM, Brenner PF, Mishell DR (November 1982). "Comparison of pharmacodynamic properties of various estrogen formulations". Am. J. Obstet. Gynecol. 144 (5): 511–8. doi:10.1016/0002-9378(82)90218-6. PMID 6291391.
- L'Hermite M (September 1990). "Risks of estrogens and progestogens". Maturitas. 12 (3): 215–46. doi:10.1016/0378-5122(90)90005-q. PMID 2170823.
- Carlström K, Pschera H, Lunell NO (December 1988). "Serum levels of oestrogens, progesterone, follicle-stimulating hormone and sex-hormone-binding globulin during simultaneous vaginal administration of 17 beta-oestradiol and progesterone in the pre- and post-menopause". Maturitas. 10 (4): 307–16. doi:10.1016/0378-5122(88)90066-7. PMID 3147360.
- Sitruk-Ware R, Nath A (June 2011). "Metabolic effects of contraceptive steroids". Rev Endocr Metab Disord. 12 (2): 63–75. doi:10.1007/s11154-011-9182-4. PMID 21538049. S2CID 23760705.
- Fruzzetti F, Trémollieres F, Bitzer J (May 2012). "An overview of the development of combined oral contraceptives containing estradiol: focus on estradiol valerate/dienogest". Gynecol. Endocrinol. 28 (5): 400–8. doi:10.3109/09513590.2012.662547. PMC 3399636. PMID 22468839.
- Mueller A, Dittrich R, Binder H, Kuehnel W, Maltaris T, Hoffmann I, Beckmann MW (July 2005). "High dose estrogen treatment increases bone mineral density in male-to-female transsexuals receiving gonadotropin-releasing hormone agonist in the absence of testosterone". Eur. J. Endocrinol. 153 (1): 107–13. doi:10.1530/eje.1.01943. PMID 15994752.
- Mueller A, Binder H, Cupisti S, Hoffmann I, Beckmann MW, Dittrich R (March 2006). "Effects on the male endocrine system of long-term treatment with gonadotropin-releasing hormone agonists and estrogens in male-to-female transsexuals". Horm. Metab. Res. 38 (3): 183–7. doi:10.1055/s-2006-925198. PMID 16673210.
- Mueller A, Zollver H, Kronawitter D, Oppelt PG, Claassen T, Hoffmann I, Beckmann MW, Dittrich R (February 2011). "Body composition and bone mineral density in male-to-female transsexuals during cross-sex hormone therapy using gonadotrophin-releasing hormone agonist". Exp. Clin. Endocrinol. Diabetes. 119 (2): 95–100. doi:10.1055/s-0030-1255074. PMID 20625973.
- Odlind V, Milsom I, Persson I, Victor A (June 2002). "Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills?". Acta Obstet Gynecol Scand. 81 (6): 482–90. doi:10.1034/j.1600-0412.2002.810603.x. PMID 12047300. S2CID 26054257.
- Kohli, M.; Alikhan, M. A.; Spencer, H. J.; Carter, G. (2004). "Phase I trial of intramuscular estradiol valerate (I/M-E) in hormone refractory prostate cancer". Journal of Clinical Oncology. 22 (14_suppl): 4726. doi:10.1200/jco.2004.22.90140.4726. ISSN 0732-183X.
- Kuhli M, McClellan J (2001). "Parenteral estrogen therapy in advanced prostate cancer: retrospective analysis of intramuscular estradiol valerate in "hormone refractory" prostate disease". Proc Am Soc Clin Oncol. 20: 2407a. ISSN 0736-7589.
- Ryan CJ, Small EJ (December 2003). "Role of secondary hormonal therapy in the management of recurrent prostate cancer". Urology. 62 Suppl 1: 87–94. doi:10.1016/j.urology.2003.10.002. PMID 14747046.
The use of parenteral estradiol [valerate] in patients with progressive disease after secondary hormonal therapy resulted in PSA decreases in 5 of 5 patients without metastatic disease in a pilot study. In this same study, parenteral estradiol in combination with chemotherapy was also administered.49
- Kohli M (January 2006). "Phase II study of transdermal estradiol in androgen-independent prostate carcinoma". Cancer. 106 (1): 234–5, author reply 235. doi:10.1002/cncr.21528. PMID 16284988. S2CID 11047031.
[...] we explored the effect of combination parenteral estrogen (intramuscular estradiol valerate) and chemotherapy on coagulation and plasminogen system activation in androgen independent stage in a Phase I trial.3 Our primary goal was to monitor coagulation and plasminogen system activation and was performed by using subclinical markers such as thrombin–antithrombin complex (TAT; reference range,1.0 – 4.1 μg/L) and quantitative D-dimer levels (QDD, range:0 – 250 ng/mL) as surrogate markers predictive for thrombohemorrhagic complications. Three escalating doses (10 mg, 20 mg, 40 mg) of intramuscular estradiol valerate were administered every 2 weeks in 12 patients. Before each estradiol valerate dose, these markers were measured, and patients with rising levels above baseline measurements were given once daily prophylaxis with 60 mg of low molecular weight heparin. We found that the majority of patients (10 of 12) had subclinical hemostatic activation as measured by rising plasma TAT and QDD levels after estradiol dosing, which returned to patient-specific baseline with daily prophylactic anticoagulation. No clinical thrombosis or hemorrhagic event was observed.
- Horský, Jan; Presl, Jiří (1981). "Hormonal Treatment of Disorders of the Menstrual Cycle". In J. Horsky; J. Presl (eds.). Ovarian Function and its Disorders: Diagnosis and Therapy. Springer Science & Business Media. pp. 309–332. doi:10.1007/978-94-009-8195-9_11. ISBN 978-94-009-8195-9.
- George Morris Piersol (1975). The Cyclopedia of Medicine, Surgery, Specialties. F. A. Davis Company.
- Wajchenberg BL (December 2000). "Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome". Endocr. Rev. 21 (6): 697–738. doi:10.1210/edrv.21.6.0415. PMID 11133069.
The androgen receptor in female adipose tissue seems to have the same characteristics as that found in male adipose tissue whereas estrogen treatment down regulates the density of this receptor (34), which might be a mechanism whereby estrogens protect adipose tissue from androgen effects. [...] The possible mechanisms for the effects of estrogens on the determination of body fat distribution include the down-regulation of the androgen receptor, thereby preventing androgen effects (34) as mentioned earlier [...]
- Brown LM, Clegg DJ (October 2010). "Central effects of estradiol in the regulation of food intake, body weight, and adiposity". J. Steroid Biochem. Mol. Biol. 122 (1–3): 65–73. doi:10.1016/j.jsbmb.2009.12.005. PMC 2889220. PMID 20035866.
[...] estrogen down-regulates AR expression in subcutaneous fat [17].
- Bjorntorp P (January 1997), "Endocrine abnormalities in obesity", Diabetes Reviews, 5 (1): 52–68, ISSN 1066-9442,
Transsexual women treated with testosterone accumulate visceral fat, but this seems to be the case only where an oophorectomy has been performed (119). [...] The observation that visceral fat accumulation occurs only in transsexual women who have had an oophorectomy (119) suggests that the remaining estrogen production before the oophorectomy (106) was protective. The androgen receptor in female adipose tissue seems to have the same characteristics as that found in male adipose tissue (85; M. Li and P. B., unpublished observations). However, estrogen treatment downregulates the density of this receptor (M. Li and P. B., unpublished observations), which might be a mechanism whereby estrogen protects adipose tissue from androgen effects. Therefore, when estrogen levels become sufficiently low, visceral fat accumulation may occur. The balance between androgens and estrogens therefore seems to be of significance; perhaps the lack of estrogen is more important than the relatively small androgen excess in hyperandrogenic women with visceral accumulation of body fat. [...] Furthermore, estrogen seems to downregulate the androgen receptor density (M. Li and P. B., unpublished observations) and may therefore prevent androgen effects. This possibility is suggested by recent observations indicating that, for androgens to promote visceral fat accumulation in women, oophorectomy seems necessary (119).
- Ottosson UB, Carlström K, Johansson BG, von Schoultz B (1986). "Estrogen induction of liver proteins and high-density lipoprotein cholesterol: comparison between estradiol valerate and ethinyl estradiol". Gynecol. Obstet. Invest. 22 (4): 198–205. doi:10.1159/000298914. PMID 3817605.
- Stanczyk, Frank Z.; Archer, David F.; Bhavnani, Bhagu R. (2013). "Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment". Contraception. 87 (6): 706–727. doi:10.1016/j.contraception.2012.12.011. ISSN 0010-7824. PMID 23375353.
- Fotherby K (August 1996). "Bioavailability of orally administered sex steroids used in oral contraception and hormone replacement therapy". Contraception. 54 (2): 59–69. doi:10.1016/0010-7824(96)00136-9. PMID 8842581.
- Victor Gomel; Malcolm G. Munro; Timothy C. Rowe (1990). Gynecology: a practical approach. Williams & Wilkins. p. 132,134. ISBN 978-0-683-03631-2.
The synthetic estrogen, ethinyl estradiol, more commonly used in oral contraceptives, has a biological activity 100 times that of the native and conjugated substances.
- Nathaniel McConaghy (21 November 2013). Sexual Behavior: Problems and Management. Springer Science & Business Media. pp. 177–. ISBN 978-1-4899-1133-9.
Meyer et al. found that ethinyl estradiol was 75 to 100 times more potent than conjugated estrogen on the basis of the doses required to lower testosterone to the adult female range, 0.1 mg of the former and 7.5 to 10 mg of the latter being necessary.
- Bruce Chabner; Dan Louis Longo (1996). Cancer Chemotherapy and Biotherapy: Principles and Practice. Lippincott-Raven Publishers. p. 186. ISBN 978-0-397-51418-2.
Piperazine estrone sulfate and micronized estradiol were equipotent with respect to increases in SHBG, [...] With respect to decreased FSH, [...] ethinyl estradiol was 80 to 200-fold more potent than was piperazine estrone sulfate. [...] The initial half-life of DES is 80 minutes, with a secondary half-life of 24 hours.222
- Tommaso Falcone; William W. Hurd (2007). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. pp. 22, 362, 388. ISBN 978-0-323-03309-1.
- Düsterberg B, Nishino Y (1982). "Pharmacokinetic and pharmacological features of oestradiol valerate". Maturitas. 4 (4): 315–24. doi:10.1016/0378-5122(82)90064-0. PMID 7169965.
- Goldzieher JW, Brody SA (1990). "Pharmacokinetics of ethinyl estradiol and mestranol". American Journal of Obstetrics and Gynecology. 163 (6 Pt 2): 2114–9. doi:10.1016/0002-9378(90)90550-Q. PMID 2256522.
- Elger W, Wyrwa R, Ahmed G, Meece F, Nair HB, Santhamma B, Killeen Z, Schneider B, Meister R, Schubert H, Nickisch K (January 2017). "Estradiol prodrugs (EP) for efficient oral estrogen treatment and abolished effects on estrogen modulated liver functions". J. Steroid Biochem. Mol. Biol. 165 (Pt B): 305–311. doi:10.1016/j.jsbmb.2016.07.008. PMID 27449818. S2CID 26650319.
- Marc A. Fritz; Leon Speroff (28 March 2012). Clinical Gynecologic Endocrinology and Infertility. Lippincott Williams & Wilkins. pp. 753–. ISBN 978-1-4511-4847-3.
- Notelovitz M (March 2006). "Clinical opinion: the biologic and pharmacologic principles of estrogen therapy for symptomatic menopause". MedGenMed. 8 (1): 85. PMC 1682006. PMID 16915215.
- Goodman MP (February 2012). "Are all estrogens created equal? A review of oral vs. transdermal therapy". J Womens Health (Larchmt). 21 (2): 161–9. doi:10.1089/jwh.2011.2839. PMID 22011208.
- Nachtigall LE, Raju U, Banerjee S, Wan L, Levitz M (2000). "Serum estradiol-binding profiles in postmenopausal women undergoing three common estrogen replacement therapies: associations with sex hormone-binding globulin, estradiol, and estrone levels". Menopause. 7 (4): 243–50. doi:10.1097/00042192-200007040-00006. ISSN 1072-3714. PMID 10914617. S2CID 3076514.
- Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A, Stege R, von Schoultz B (1989). "A comparison of androgen status in patients with prostatic cancer treated with oral and/or parenteral estrogens or by orchidectomy". Prostate. 14 (2): 177–82. doi:10.1002/pros.2990140210. PMID 2523531. S2CID 25516937.
- Scarabin PY (December 2014). "Hormones and venous thromboembolism among postmenopausal women". Climacteric. 17 Suppl 2: 34–7. doi:10.3109/13697137.2014.956717. PMID 25223916. S2CID 5084606.
- Mohammed K, Abu Dabrh AM, Benkhadra K, Al Nofal A, Carranza Leon BG, Prokop LJ, Montori VM, Faubion SS, Murad MH (November 2015). "Oral vs Transdermal Estrogen Therapy and Vascular Events: A Systematic Review and Meta-Analysis". J. Clin. Endocrinol. Metab. 100 (11): 4012–20. doi:10.1210/jc.2015-2237. PMID 26544651.
- Bińkowska M (October 2014). "Menopausal hormone therapy and venous thromboembolism". Prz Menopauzalny. 13 (5): 267–72. doi:10.5114/pm.2014.46468. PMC 4520375. PMID 26327865.
- S. Campbell (6 December 2012). The Management of the Menopause & Post-Menopausal Years: The Proceedings of the International Symposium held in London 24–26 November 1975 Arranged by the Institute of Obstetrics and Gynaecology, The University of London. Springer Science & Business Media. pp. 395–. ISBN 978-94-011-6165-7.
- Phillips I, Shah SI, Duong T, Abel P, Langley RE (2014). "Androgen Deprivation Therapy and the Re-emergence of Parenteral Estrogen in Prostate Cancer". Oncol Hematol Rev. 10 (1): 42–47. doi:10.17925/ohr.2014.10.1.42. PMC 4052190. PMID 24932461.
- Henriksson P, Carlström K, Pousette A, Gunnarsson PO, Johansson CJ, Eriksson B, Altersgård-Brorsson AK, Nordle O, Stege R (1999). "Time for revival of estrogens in the treatment of advanced prostatic carcinoma? Pharmacokinetics, and endocrine and clinical effects, of a parenteral estrogen regimen". Prostate. 40 (2): 76–82. doi:10.1002/(sici)1097-0045(19990701)40:2<76::aid-pros2>3.0.co;2-q. PMID 10386467. S2CID 12240276.
- Russell N, Cheung A, Grossmann M (August 2017). "Estradiol for the mitigation of adverse effects of androgen deprivation therapy". Endocr. Relat. Cancer. 24 (8): R297–R313. doi:10.1530/ERC-17-0153. PMID 28667081.
- Shellenberger, T. E. (1986). "Pharmacology of estrogens". The Climacteric in Perspective. pp. 393–410. doi:10.1007/978-94-009-4145-8_36. ISBN 978-94-010-8339-3.
Ethinyl estradiol is a synthetic and comparatively potent estrogen. As a result of the alkylation in 17-C position it is not a substrate for 17β dehydrogenase, an enzyme which transforms natural estradiol-17β to the less potent estrone in target organs.
- Mueck AO, Seeger H, Rabe T (December 2010). "Hormonal contraception and risk of endometrial cancer: a systematic review". Endocr. Relat. Cancer. 17 (4): R263–71. doi:10.1677/ERC-10-0076. PMID 20870686.
Further reading
- Alfred S. Wolf; H.P.G. Schneider (1989). Östrogene in Diagnostik und Therapie [Estrogens in Diagnostics and Therapy]. Springer-Verlag. pp. 1–. ISBN 978-3-642-75101-1.
- Kuhl H (September 1990). "Pharmacokinetics of oestrogens and progestogens". Maturitas. 12 (3): 171–97. doi:10.1016/0378-5122(90)90003-O. PMID 2170822.
- Lobo RA, Cassidenti DL (January 1992). "Pharmacokinetics of oral 17 beta-estradiol". J Reprod Med. 37 (1): 77–84. PMID 1548642.
- O'Connell MB (September 1995). "Pharmacokinetic and pharmacologic variation between different estrogen products". J Clin Pharmacol. 35 (9S): 18S–24S. doi:10.1002/j.1552-4604.1995.tb04143.x. PMID 8530713. S2CID 10159196.
- Michael Oettel; Ekkehard Schillinger (1999). Estrogens and Antiestrogens I: Physiology and Mechanisms of Action of Estrogens and Antiestrogens. Springer Science & Business Media. ISBN 978-3-642-58616-3.
- Michael Oettel; Ekkehard Schillinger (1999). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen. Springer Science & Business Media. ISBN 978-3-642-60107-1.
- Ruggiero RJ, Likis FE (2002). "Estrogen: physiology, pharmacology, and formulations for replacement therapy". J Midwifery Womens Health. 47 (3): 130–8. doi:10.1016/S1526-9523(02)00233-7. PMID 12071379.
- Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration" (PDF). Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947. S2CID 24616324.
- Barnes, Randall B.; Levrant, Seth G. (2007). Treatment of the Postmenopausal Woman. pp. 767–777. doi:10.1016/B978-012369443-0/50066-1. ISBN 9780123694430.
- Fruzzetti F, Trémollieres F, Bitzer J (2012). "An overview of the development of combined oral contraceptives containing estradiol: focus on estradiol valerate/dienogest". Gynecol. Endocrinol. 28 (5): 400–8. doi:10.3109/09513590.2012.662547. PMC 3399636. PMID 22468839.
- Stanczyk FZ, Archer DF, Bhavnani BR (2013). "Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment". Contraception. 87 (6): 706–27. doi:10.1016/j.contraception.2012.12.011. PMID 23375353.