Reperfusion injury
| Reperfusion injury | |
|---|---|
| Other names | Reperfusion insult |
| Specialty | Cardiology |
Reperfusion injury, sometimes called ischemia-reperfusion injury (IRI) or reoxygenation injury, is the tissue damage caused when blood supply returns to tissue (re- + perfusion) after a period of ischemia or lack of oxygen (anoxia or hypoxia). The absence of oxygen and nutrients from blood during the ischemic period creates a condition in which the restoration of circulation results in inflammation and oxidative damage through the induction of oxidative stress rather than (or along with) restoration of normal function.
Reperfusion injury is distinct from cerebral hyperperfusion syndrome (sometimes called "Reperfusion syndrome"), a state of abnormal cerebral vasodilation.
Mechanisms
Reperfusion of ischemic tissues is often associated with microvascular injury, particularly due to increased permeability of capillaries and arterioles that lead to an increase of diffusion and fluid filtration across the tissues. Activated endothelial cells produce more reactive oxygen species but less nitric oxide following reperfusion, and the imbalance results in a subsequent inflammatory response.[1] The inflammatory response is partially responsible for the damage of reperfusion injury. White blood cells, carried to the area by the newly returning blood, release a host of inflammatory factors such as interleukins as well as free radicals in response to tissue damage.[2] The restored blood flow reintroduces oxygen within cells that damages cellular proteins, DNA, and the plasma membrane. Damage to the cell's membrane may in turn cause the release of more free radicals. Such reactive species may also act indirectly in redox signaling to turn on apoptosis. White blood cells may also bind to the endothelium of small capillaries, obstructing them and leading to more ischemia.[2] Another hypothesis would be that normally, tissues contain free radical scavengers to avoid damage by oxidizing species normally contained in the blood. Ischemic tissue would have decreased function of these scavengers because of cell injury. Once blood flow is reestablished, oxygen species contained in the blood will damage the ischemic tissue because the function of the scavengers is decreased.
Reperfusion injury plays a major part in the biochemistry of hypoxic brain injury in stroke. Similar failure processes are involved in brain failure following reversal of cardiac arrest;[3] control of these processes is the subject of ongoing research. Repeated bouts of ischemia and reperfusion injury also are thought to be a factor leading to the formation and failure to heal of chronic wounds such as pressure sores and diabetic foot ulcer.[4] Continuous pressure limits blood supply and causes ischemia, and the inflammation occurs during reperfusion. As this process is repeated, it eventually damages tissue enough to cause a wound.[4]
The main reason for the acute phase of ischemia-reperfusion injury is oxygen deprivation and, therefore, arrest of generation of ATP (cellular energy currency) by mitochondria oxidative phosphorylation. Tissue damage due to the general energy deficit during ischemia is followed by reperfusion (increase of oxygen level) when the injury is enhanced. Mitochondrial complex I is thought to be the most vulnerable enzyme to tissue ischemia/reperfusion but the mechanism of damage is different in different tissues. For example brain ischemia/reperfusion injury is mediated via complex I redox-dependent inactivation.[5] It was found that lack of oxygen leads to conditions in which mitochondrial complex I loses its natural cofactor, flavin mononucleotide (FMN) and become inactive.[6] When oxygen is present the enzyme catalyzes a physiological reaction of NADH oxidation by ubiquinone, supplying electrons downstream of the respiratory chain (complexes III and IV). Ischemia leads to dramatic increase of succinate level.[7] In the presence of succinate mitochondria catalyze reverse electron transfer so that fraction of electrons from succinate is directed upstream to FMN of complex I.[8] Reverse electron transfer results in a reduction of complex I FMN, increased generation of ROS, followed by a loss of the reduced cofactor (FMNH2) and impairment of mitochondria energy production.[8] The FMN loss by complex I and I/R injury can be alleviated by the administration of FMN precursor, riboflavin.[6]
In prolonged ischemia (60 minutes or more), hypoxanthine is formed as a breakdown product of ATP metabolism. The enzyme xanthine dehydrogenase acts in reverse, that is as a xanthine oxidase as a result of the higher availability of oxygen. This oxidation results in molecular oxygen being converted into highly reactive superoxide and hydroxyl radicals. Xanthine oxidase also produces uric acid, which may act as both a prooxidant and as a scavenger of reactive species such as peroxynitrite. Excessive nitric oxide produced during reperfusion reacts with superoxide to produce the potent reactive species peroxynitrite. Such radicals and reactive oxygen species attack cell membrane lipids, proteins, and glycosaminoglycans, causing further damage. They may also initiate specific biological processes by redox signaling.
Reperfusion can cause hyperkalemia.[9]
Reperfusion injury is a primary concern in liver transplantation surgery.[10]
Treatment
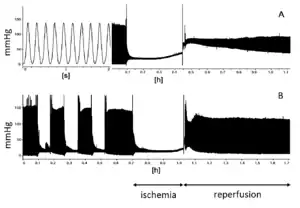
A study of aortic cross-clamping, a common procedure in cardiac surgery, demonstrated a strong potential benefit with further research ongoing.
Therapeutic hypothermia
An intriguing area of research demonstrates the ability of a reduction in body temperature to limit ischemic injuries. This procedure is called therapeutic hypothermia, and it has been shown by a number of large, high-quality randomised trials to significantly improve survival and reduce brain damage after birth asphyxia in newborn infants, almost doubling the chance of normal survival. For a full review see Hypothermia therapy for neonatal encephalopathy.
However, the therapeutic effect of hypothermia does not confine itself to metabolism and membrane stability. Another school of thought focuses on hypothermia's ability to prevent the injuries that occur after circulation returns to the brain, or what is termed reperfusion injuries. In fact an individual suffering from an ischemic insult continues suffering injuries well after circulation is restored. In rats it has been shown that neurons often die a full 24 hours after blood flow returns. Some theorize that this delayed reaction derives from the various inflammatory immune responses that occur during reperfusion.[11] These inflammatory responses cause intracranial pressure, pressure which leads to cell injury and in some situations cell death. Hypothermia has been shown to help moderate intracranial pressure and therefore to minimize the harmful effect of a patient's inflammatory immune responses during reperfusion. Beyond this, reperfusion also increases free radical production. Hypothermia too has been shown to minimize a patient's production of deadly free radicals during reperfusion. Many now suspect it is because hypothermia reduces both intracranial pressure and free radical production that hypothermia improves patient outcome following a blockage of blood flow to the brain.[12]
Hydrogen sulfide treatment
There are some preliminary studies in mice that seem to indicate that treatment with hydrogen sulfide (H2S) can have a protective effect against reperfusion injury.[13]
Cyclosporin
In addition to its well-known immunosuppressive capabilities, the one-time administration of cyclosporin at the time of percutaneous coronary intervention (PCI) has been found to deliver a 40 percent reduction in infarct size in a small group proof of concept study of human patients with reperfusion injury published in The New England Journal of Medicine in 2008.[14]
Cyclosporin has been confirmed in studies to inhibit the actions of cyclophilin D, a protein which is induced by excessive intracellular calcium flow to interact with other pore components and help open the MPT pore. Inhibiting cyclophilin D has been shown to prevent the opening of the MPT pore and protect the mitochondria and cellular energy production from excessive calcium inflows.[15]
However, the studies CIRCUS and CYCLE (published in September 2015 and February 2016 respectively) looked at the use of cyclosporin as a one time IV dose given right before perfusion therapy (PCI). Both studies found there is no statistical difference in outcome with cyclosporin administration.[16][17] Research is ongoing.
Reperfusion leads to biochemical imbalances within the cell that lead to cell death and increased infarct size. More specifically, calcium overload and excessive production of reactive oxygen species in the first few minutes after reperfusion set off a cascade of biochemical changes that result in the opening of the so-called mitochondrial permeability transition pore (MPT pore) in the mitochondrial membrane of cardiac cells.[15]
The opening of the MPT pore leads to the inrush of water into the mitochondria, resulting in mitochondrial dysfunction and collapse. Upon collapse, the calcium is then released to overwhelm the next mitochondria in a cascading series of events that cause mitochondrial energy production supporting the cell to be reduced or stopped completely. The cessation of energy production results in cellular death. Protecting mitochondria is a viable cardioprotective strategy.[18]
In 2008, an editorial in the New England Journal of Medicine called for more studies to determine if cyclosporin can become a treatment to ameliorate reperfusion injury by protecting mitochondria.[18] To that end, in 2011 the researchers involved in the original 2008 NEJM study initiated a phase III clinical study of reperfusion injury in 1000 myocardial infarction patients in centers throughout Europe. Results of that study were announced in 2015 and indicated that "intravenous ciclosporin did not result in better clinical outcomes than those with placebo and did not prevent adverse left ventricular remodeling at 1 year".[16] This same process of mitochondrial destruction through the opening of the MPT pore is implicated in making traumatic brain injuries much worse.[19] Ciclosporin is currently in a phase II/III (adaptive) clinical study in Europe to determine its ability to ameliorate neuronal cellular damage in traumatic brain injury.
TRO40303
TRO40303 is a new cardioprotective compound that was shown to inhibit the MPT pore and reduce infarct size after ischemia-reperfusion. It was developed by Trophos company and currently is in Phase I clinical trial.[20]
Stem cell therapy
Recent investigations suggest a possible beneficial effect of mesenchymal stem cells on heart and kidney reperfusion injury.[21][22]
Superoxide dismutase
Superoxide dismutase is an effective anti-oxidant enzyme which converts superoxide anions to water and hydrogen peroxide. Recent researches have shown significant therapeutic effects on pre-clinical models of reperfusion injury after ischemic stroke.[23][24]
Metformin
A series of 2009 studies published in the Journal of Cardiovascular Pharmacology suggest that Metformin may prevent cardiac reperfusion injury by inhibition of Mitochondrial Complex I and the opening of MPT pore and in rats.[25][26]
Riboflavin
In neonatal in vivo model of brain ischemia/reperfusion, tissue injury can be alleviated by the administration of FMN precursor, riboflavin that prevents inactivation of mitochondrial complex I.[6][27]
Cannabinoids
A study published in 2012 show that the synthetic analogue of the phytocannabinoid Tetrahydrocannabivarin (THCV), Δ8-Tetrahydrocannabivarin (Δ8-THCV) and its metabolite 11-OH-Δ8-THCV, prevent hepatic ischaemia/reperfusion injury by decreasing oxidative stress and inflammatory responses through cannabinoid CB2 receptors and thereby decrease tissue injury and inflammation with a protective effect against liver damage. Pretreatment with a CB2 receptor antagonist attenuated the protective effects of Δ8-THCV, while a CB1 antagonist tended to enhance it.[28]
An earlier study published in 2011 found, that Cannabidiol (CBD) also protects against hepatic ischemia/reperfusion injury by attenuating inflammatory signaling and response of oxidative and nitrative stress, and thereby cell death and tissue injury, but independent from classical CB1 and CB2 receptors.[29]
Reperfusion protection in obligate hibernators
Obligatory hibernators such as the ground squirrels show resistance to ischemia/reperfusion (I/R) injury in liver, heart, and small intestine during the hibernation season when there is a switch from carbohydrate metabolism to lipid metabolism for cellular energy supply.[30][31][32] This metabolic switch limits anaerobic metabolism and the formation of lactate, a herald of poor prognosis and multi-organ failure (MOF) after I/R injury. In addition, the increase in lipid metabolism generates ketone bodies and activates peroxisome proliferating-activated receptors (PPARs), both of which have been shown to be protective against I/R injury.[33]
See also
References
- ↑ Carden, DL; Granger, DN (Feb 2000). "Pathophysiology of ischaemia-reperfusion injury". The Journal of Pathology. 190 (3): 255–66. doi:10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. PMID 10685060.
- 1 2 Clark, Wayne M. (January 5, 2005). "Reperfusion Injury in Stroke". eMedicine. WebMD. Retrieved 2006-08-09.
- ↑ Crippen, David. "Brain Failure and Brain Death: Introduction". Scientific American Surgery, Critical Care, April 2005. Retrieved 2007-01-09.
- 1 2 Mustoe T. (2004). "Understanding chronic wounds: a unifying hypothesis on their pathogenesis and implications for therapy". American Journal of Surgery. 187 (5A): 65S–70S. doi:10.1016/S0002-9610(03)00306-4. PMID 15147994.
- ↑ Galkin, A. (2019). "Brain Ischemia/Reperfusion Injury and Mitochondrial Complex I Damage". Biochemistry. Biokhimiia. 84 (11): 1411–1423. doi:10.1134/S0006297919110154. ISSN 1608-3040. PMID 31760927.
- 1 2 3 Stepanova, Anna; Sosunov, Sergey; Niatsetskaya, Zoya; Konrad, Csaba; Starkov, Anatoly A.; Manfredi, Giovanni; Wittig, Ilka; Ten, Vadim; Galkin, Alexander (2019-09-20). "Redox-Dependent Loss of Flavin by Mitochondrial Complex I in Brain Ischemia/Reperfusion Injury". Antioxidants & Redox Signaling. 31 (9): 608–622. doi:10.1089/ars.2018.7693. ISSN 1557-7716. PMC 6657304. PMID 31037949.
- ↑ Sahni, Prateek V.; Zhang, Jimmy; Sosunov, Sergey; Galkin, Alexander; Niatsetskaya, Zoya; Starkov, Anatoly; Brookes, Paul S.; Ten, Vadim S. (2018). "Krebs cycle metabolites and preferential succinate oxidation following neonatal hypoxic-ischemic brain injury in mice". Pediatric Research. 83 (2): 491–497. doi:10.1038/pr.2017.277. ISSN 1530-0447. PMC 5866163. PMID 29211056.
- 1 2 Stepanova, Anna; Kahl, Anja; Konrad, Csaba; Ten, Vadim; Starkov, Anatoly S.; Galkin, Alexander (2017). "Reverse electron transfer results in a loss of flavin from mitochondrial complex I: Potential mechanism for brain ischemia reperfusion injury". Journal of Cerebral Blood Flow and Metabolism. 37 (12): 3649–3658. doi:10.1177/0271678X17730242. ISSN 1559-7016. PMC 5718331. PMID 28914132.
- ↑ John L. Atlee (2007). Complications in anesthesia. Elsevier Health Sciences. pp. 55–. ISBN 978-1-4160-2215-2. Retrieved 25 July 2010.
- ↑ Lemasters JJ. (1997). "Reperfusion injury after liver preservation for transplantation". Annual Review of Pharmacology and Toxicology. 37: 327–38. doi:10.1146/annurev.pharmtox.37.1.327. PMID 9131256.
- ↑ Adler, Jerry. "Back From the Dead." Newsweek. July 23, 2007.
- ↑ Polderman KH (2004). "Application of therapeutic hypothermia in the ICU: opportunities and pitfalls of a promising treatment modality. Part 1: Indications and evidence". Intensive Care Med. 30 (4): 556–75. doi:10.1007/s00134-003-2152-x. PMID 14767591.
- ↑ Elrod J.W., J.W. Calvert, M.R. Duranski, D.J. Lefer. "Hydrogen sulfide donor protects against acute myocardial ischemia-reperfusion injury." Circulation 114(18):II172, 2006.
- ↑ Piot C.; Croiselle P.; Staat P.; et al. (2008). "Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infaction". New England Journal of Medicine. 359 (5): 473–481. doi:10.1056/nejmoa071142. PMID 18669426.
- 1 2 Javadov S.; Karmazyn M. (2007). "Mitochondrial Permeability Transition Pore Opening as an Endpoint to Initiate Cell Death and as a Putative Target for Cardioprotection". Cell Physiol Biochem. 20 (1–4): 1–22. doi:10.1159/000103747. PMID 17595511.
- 1 2 Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, et al. (2015). "Cyclosporine before PCI in Patients with Acute Myocardial Infarction" (PDF). New England Journal of Medicine. 373 (11): 1021–31. doi:10.1056/NEJMoa1505489. hdl:10044/1/41761. PMID 26321103.
- ↑ Ottani F, Latini R, Staszewsky L, La Vecchia L, Locuratolo N, Sicuro M, Masson S, Barlera S, Milani V, Lombardi M, Costalunga A, Mollichelli N, Santarelli A, De Cesare N, Sganzerla P, Boi A, Maggioni AP, Limbruno U (2016). "Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial". J. Am. Coll. Cardiol. 67 (4): 365–374. doi:10.1016/j.jacc.2015.10.081. PMID 26821623.
- 1 2 Hausenloy D.; Yellon D. (2008). "Time to take myocardial reperfusion injury seriously". New England Journal of Medicine. 359 (5): 518–520. doi:10.1056/nejme0803746. PMID 18669431.
- ↑ Sullivan PG, Sebastian AH, Hall ED (2011). "Therapeutic window analysis of the neuroprotective effects of cyclosporine A after traumatic brain injury". J. Neurotrauma. 28 (2): 311–8. doi:10.1089/neu.2010.1646. PMC 3037811. PMID 21142667.
- ↑ Le Lamer S (Feb 2014). "Translation of TRO40303 from myocardial infarction models to demonstration of safety and tolerance in a randomized Phase I trial". J Transl Med. 12: 38. doi:10.1186/1479-5876-12-38. PMC 3923730. PMID 24507657.
- ↑ van der Spoel TI (Sep 2011). "Human relevance of pre-clinical studies in stem cell therapy: systematic review and meta-analysis of large animal models of ischaemic heart disease". Cardiovasc Res. 91 (4): 649–58. doi:10.1093/cvr/cvr113. PMID 21498423.
- ↑ Zhao JJ (Jan 2014). "Protection of mesenchymal stem cells on acute kidney injury". Mol Med Rep. 9 (1): 91–96. doi:10.3892/mmr.2013.1792. PMID 24220681.
- ↑ Jiang, Yuhang; Arounleut, Phonepasong; Rheiner, Steven; Bae, Younsoo; Kabanov, Alexander V.; Milligan, Carol; Manickam, Devika S. (2016-06-10). "SOD1 nanozyme with reduced toxicity and MPS accumulation". Journal of Controlled Release. Thirteenth International Nanomedicine and Drug Delivery Symposium. 231: 38–49. doi:10.1016/j.jconrel.2016.02.038. PMID 26928528.
- ↑ Jiang, Yuhang; Brynskikh, Anna M.; S-Manickam, Devika; Kabanov, Alexander V. (2015-09-10). "SOD1 nanozyme salvages ischemic brain by locally protecting cerebral vasculature". Journal of Controlled Release. 213: 36–44. doi:10.1016/j.jconrel.2015.06.021. PMC 4684498. PMID 26093094.
- ↑ Paiva, Marta; Riksen, Niels P.; Davidson, Sean M.; Hausenloy, Derek J.; Monteiro, Pedro; Gonçalves, Lino; Providência, Luís; Rongen, Gerard A.; Smits, Paul (2009-05-01). "Metformin prevents myocardial reperfusion injury by activating the adenosine receptor". Journal of Cardiovascular Pharmacology. 53 (5): 373–378. doi:10.1097/FJC.0b013e31819fd4e7. ISSN 1533-4023. PMID 19295441.
- ↑ Bhamra, Gurpreet S.; Hausenloy, Derek J.; Davidson, Sean M.; Carr, Richard D.; Paiva, Marta; Wynne, Abigail M.; Mocanu, Mihaela M.; Yellon, Derek M. (2008-05-01). "Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening". Basic Research in Cardiology. 103 (3): 274–284. doi:10.1007/s00395-007-0691-y. ISSN 0300-8428. PMID 18080084.
- ↑ Ten, Vadim; Galkin, Alexander (2019). "Mechanism of mitochondrial complex I damage in brain ischemia/reperfusion injury. A hypothesis". Molecular and Cellular Neurosciences. 100: 103408. doi:10.1016/j.mcn.2019.103408. ISSN 1095-9327. PMID 31494262.
- ↑ Bátkai, Sándor; Mukhopadhyay, Partha; Horváth, Bėla; Rajesh, Mohanraj; Gao, Rachel Y; Mahadevan, Anu; Amere, Mukkanti; Battista, Natalia; Lichtman, Aron H (2012). "Δ8-Tetrahydrocannabivarin prevents hepatic ischaemia/reperfusion injury by decreasing oxidative stress and inflammatory responses through cannabinoid CB2 receptors". British Journal of Pharmacology. 165 (8): 2450–2461. doi:10.1111/j.1476-5381.2011.01410.x. ISSN 0007-1188. PMC 3423240. PMID 21470208.
- ↑ Mukhopadhyay, Partha; Rajesh, Mohanraj; Horváth, Béla; Bátkai, Sándor; Park, Ogyi; Tanashian, Galin; Gao, Rachel Y; Patel, Vivek; Wink, David A. (2011-05-15). "Cannabidiol protects against hepatic ischemia/reperfusion injury by attenuating inflammatory signaling and response, oxidative/nitrative stress, and cell death". Free Radical Biology & Medicine. 50 (10): 1368–1381. doi:10.1016/j.freeradbiomed.2011.02.021. ISSN 0891-5849. PMC 3081988. PMID 21362471.
- ↑ Dark, J (2005). "Annual lipid cycles in hibernators: integration of physiology and behavior". Annual Review of Nutrition. 25: 469–97. doi:10.1146/annurev.nutr.25.050304.092514. PMID 16011475.
- ↑ Andrews, MT (May 2007). "Advances in molecular biology of hibernation in mammals". BioEssays. 29 (5): 431–40. doi:10.1002/bies.20560. PMID 17450592.
- ↑ Kurtz, CC; Lindell, SL; Mangino, MJ; Carey, HV (November 2006). "Hibernation confers resistance to intestinal ischemia-reperfusion injury". American Journal of Physiology. Gastrointestinal and Liver Physiology. 291 (5): G895–901. doi:10.1152/ajpgi.00155.2006. PMID 16751173.
- ↑ Zingarelli, B; Hake, PW; O'Connor, M; Burroughs, TJ; Wong, HR; Solomkin, JS; Lentsch, AB (June 2009). "Lung injury after hemorrhage is age dependent: role of peroxisome proliferator-activated receptor gamma". Critical Care Medicine. 37 (6): 1978–87. doi:10.1097/CCM.0b013e31819feb4d. PMC 2765201. PMID 19384226.
External links
| Authority control: National libraries |
|---|