Primary sclerosing cholangitis
| Primary sclerosing cholangitis | |
|---|---|
| Other names: Stenosing cholangitis, fibrosing cholangitis, chronic obliterative cholangitis | |
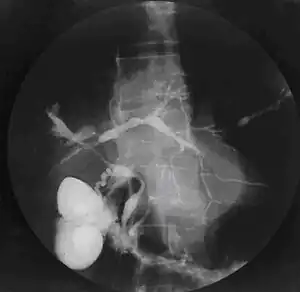 | |
| Cholangiogram of primary sclerosing cholangitis. | |
| Specialty | Gastroenterology |
| Symptoms | Yellow discoloration, itching, right upper abdominal pain[1] |
| Complications | Liver failure, gallbladder cancer, bile duct cancer, bile duct infection[2] |
| Usual onset | 30 to 50 years old[2] |
| Duration | Long-term[2] |
| Types | Classic, small-duct, associated with autoimmune hepatitis[1] |
| Causes | Unknown[1] |
| Risk factors | Inflammatory bowel disease, family history[2] |
| Diagnostic method | Elevated alkaline phosphatase, MRCP, ERCP, liver biopsy[1] |
| Differential diagnosis | Autoimmune hepatitis, pancreatitis, primary biliary cirrhosis, common bile duct stones[1] |
| Treatment | Immunosuppressants, cholestyramine, liver transplant[1] |
| Prognosis | Life expectancy 10 years without transplant[1] |
| Frequency | 11 per 100,000 people[2] |
Primary sclerosing cholangitis (PSC) is a long-term disease in which the bile ducts inside and outside the liver become inflamed and scarred, and are eventually narrowed or blocked.[2] Many people initially have no symptoms; however over time may develop liver disease with yellow discoloration of the skin and eyes, itching, and right upper abdominal pain.[1] Complications may include liver failure, gallbladder cancer, bile duct cancer, bile duct infection, and fat soluble vitamin deficiencies.[2][1]
The cause is unknown.[1] In approximately 70% of cases the person has inflammatory bowel disease (IBD), most often ulcerative colitis (UC) and conversely about 7.5% of people with UC have PSC.[2][1] Other risk factors include a family history of the disease and being a nonsmoker.[1] The underlying mechanism is believed to be that of an autoimmune disease.[2] Diagnosis may be made based a persistently elevated alkaline phosphatase and MRCP after ruling out other possible causes.[1]
There is no cure.[1] Efforts to slow the disease may include the use of immunosuppressants.[1] Cholestyramine or naltrexone may be used to improve itchiness.[1] A liver transplant is eventually required in about 40% of people, and despite transplantation the disease recurs in 25% of cases.[3] Without a liver transplant the typical life expectancy is 10 years.[1]
PSC is a rare, affecting about 11 per 100,000 people.[2] Onset is usually in peoples 30s or 40s.[2] People of Northern European ancestry are more commonly affected than those of Asian descent. Males are affected twice as often as females.[2][1]The disease was first described in the mid-1800s but was not fully characterized until the 1970s.[4]
Signs and symptoms

Nearly half of people with PSC do not have symptoms and are often incidentally discovered to have PSC due to abnormal liver function tests,[3] but a substantial proportion will have debilitating signs and symptoms of the disease.[5] Signs and symptoms of PSC may include severe itching and non-specific fatigue. Yellowing of the skin and white portion of the eyes may also be seen. Enlargement of the liver and spleen are seen in approximately 40% of affected individuals. Abdominal pain affects about 20% of people with PSC.
Multiple episodes of life-threatening acute cholangitis (infection within the bile ducts) can be seen due to impaired drainage of the bile ducts, which increases the risk of infection.[6]
- Dark urine due to excess conjugated bilirubin, which is water-soluble and excreted by the kidneys (i.e. choluria)
- Malabsorption, especially of fat, and steatorrhea (fatty stool), due to an inadequate amount of bile reaching the small intestine, leading to decreased levels of the fat-soluble vitamins, A, D, E, and K.
- Portal hypertension, a complication of cirrhosis, which can manifest with esophageal and parastomal varices[7] as well as hepatic encephalopathy (mental status alteration/disturbance caused by liver dysfunction and shunting of blood away from the scarred liver; such that ammonia detoxification is reduced with concomitant encephalopathy).
Cause
The exact cause of primary sclerosing cholangitis is unknown and its pathogenesis is poorly understood.[3] Although PSC is thought to be an autoimmune disease, it does not demonstrate a clear response to immunosuppressants. Thus, many experts believe it to be a complex, multifactorial (including immune-mediated) disorder and perhaps one that encompasses several different hepatobiliary diseases.[8][9]
In addition, there are longstanding, well-recognized associations between PSC and human leukocyte antigen (HLA) alleles (A1, B8, and DR3).[10]
Pathophysiology
PSC is characterized by inflammation of the bile ducts (cholangitis) with consequent stricturing (i.e., narrowing) and hardening (sclerosis) of these ducts due to scar formation, be it inside and/or outside the liver.[11] The resulting scarring of the bile ducts obstructs the flow of bile, which further perpetuates bile duct and liver injury. Chronic impairment of bile flow due to blockage and dysfunctional bile transport (cholestasis) causes progressive biliary fibrosis and ultimately biliary cirrhosis and liver failure.[12]
The primary physiological function of bile is to assist in the breakdown and absorption of fat in the intestinal tract; a relative deficiency of bile can lead to fat malabsorption and deficiencies of fat-soluble vitamins (A, D, E, K).
Liver enlargement is seen due to portal hypertension caused by compression of portal veins by the proximate sclerosed intrahepatic bile ducts, and leads to right upper quadrant abdominal pain.
Data has suggested an important association between the intestinal microbiota and PSC[13][14][15] and a process referred to as cellular senescence and the senescence-associated secretory phenotype (SASP) in the pathogenesis of PSC.[16][17]
Diagnosis
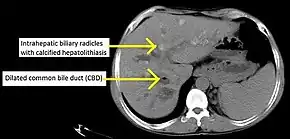
PSC is generally diagnosed on the basis of having at least two of three clinical criteria after secondary causes of sclerosing cholangitis have been ruled out:
- serum alkaline phosphatase (ALP) > 1.5x the upper limit of normal for longer than 6 months;
- cholangiography demonstrating biliary strictures or irregularity consistent with PSC; and,
- liver biopsy consistent with PSC (if available).
Historically, a cholangiogram would be obtained via endoscopic retrograde cholangiopancreatography (ERCP), which typically reveals "beading" (alternating strictures and dilation) of the bile ducts inside and/or outside the liver. Currently, the preferred option for diagnostic cholangiography, given its non-invasive yet highly accurate nature, is magnetic resonance cholangiopancreatography (MRCP), a magnetic resonance imaging technique. MRCP has unique strengths, including high spatial resolution, and can even be used to visualize the biliary tract of small animal models of PSC.[18]
Most people with PSC have evidence of autoantibodies and abnormal immunoglobulin levels.[19] For example, approximately 80% of people with PSC have perinuclear anti-neutrophil cytoplasmic antibodies (P-ANCA); however, this and other immunoglobulin findings are not specific to those with PSC and are of unclear clinical significance/consequence. Antinuclear antibodies and anti-smooth muscle antibody are found in 20%-50% of PSC patients and, likewise, are not specific for the disease but may identify a subgroup of PSC patients who also have autoimmune hepatitis (i.e. PSC-AIH overlap syndrome).[10]
Other markers which may be measured and monitored are a complete blood count, serum liver enzymes, bilirubin levels (usually grossly elevated), kidney function, and electrolytes. Fecal fat measurement is occasionally ordered when symptoms of malabsorption (e.g., gross steatorrhea) are prominent.
The differential diagnosis can include primary biliary cholangitis (formerly referred to as primary biliary cirrhosis), drug-induced cholestasis, cholangiocarcinoma, IgG4-related disease, post-liver transplantation non-anastomotic biliary strictures,[20] and HIV-associated cholangiopathy.[21] Primary sclerosing cholangitis and primary biliary cholangitis are distinct entities and exhibit important differences, including the site of tissue damage within the liver, associations with inflammatory bowel disease (IBD), which includes ulcerative colitis and Crohn's disease, response to treatment, and risks of disease progression.[22]
Classification
Primary sclerosing cholangitis is typically classified into three subgroups based on whether the small and/or large bile ducts are affected. The subgroups of PSC include the following:[3]
- Classic PSC
- Small-duct PSC
- PSC associated with autoimmune hepatitis
Management
No medication is approved for PSC in the USA. Some experts recommend a trial of ursodeoxycholic acid (UDCA), a bile acid occurring naturally in small quantities in humans, as it has been shown to lower elevated liver enzyme numbers in patients with PSC and has proven effective in other cholestatic liver diseases. However, UDCA has yet to be shown to clearly lead to improved liver histology and survival.[5][23] Guidelines from the American Association for the Study of Liver Diseases and the American College of Gastroenterology do not support the use of UDCA but guidelines from the European Association for the Study of the Liver do endorse the use of moderate doses (13-15 milligrams per kilogram) of UDCA for PSC.[3][24][25][26]
Supportive treatment for symptoms is the cornerstone of management. These therapies are aimed at relieving symptoms such as itching with antipruritics (e.g. bile acid sequestrants such as cholestyramine); antibiotics to treat episodes of ascending cholangitis; and vitamin supplements, as people with PSC are often deficient in fat-soluble vitamins (vitamin A, vitamin D, vitamin E, and vitamin K).[27]
ERCP and specialized techniques may also be needed to help distinguish between a benign PSC stricture and a bile duct cancer (cholangiocarcinoma).[28]
Liver transplantation is the only proven long-term treatment. Indications for transplantation include recurrent bacterial ascending cholangitis, decompensated cirrhosis, hepatocellular carcinoma, hilar cholangiocarcinoma, and complications of portal hypertension. Not all people are candidates for liver transplantation, and some will experience disease recurrence afterward.[8]
Prognosis
Estimated median survival from diagnosis until liver transplant or PSC-related death is 21.3 years.[29] Various models have been developed to help predict survival, but their use is generally best suited for research and not clinical purposes. A serum alkaline phosphatase less than 1.5 times the upper limit of normal has been associated with better outcomes but its utility in predicting long-term outcomes is unclear.[3]
Related diseases
The development of any of the cancers associated with PSC predicts a poor prognosis. Complications from PSC-associated cancers account for 40% of deaths from PSC.[30] Primary sclerosing cholangitis is one of the major known risk factors for cholangiocarcinoma,[31] a cancer of the biliary tree, for which the lifetime risk among patients with PSC is 10-15%.[32] This represents a 400-fold greater risk of developing cholangiocarcinoma compared to the general population.[3] Surveillance for cholangiocarcinoma in patients with PSC is encouraged, with some experts recommending annual surveillance with a specialized imaging study and serum markers,[33] although consensus regarding the modality and interval has yet to be established. Similarly, a screening colonoscopy is recommended in people who receive a new diagnosis of primary sclerosing cholangitis since their risk of colorectal cancer is 10 times higher than that of the general population.[3]
PSC is strongly associated with inflammatory bowel disease (IBD), in particular ulcerative colitis (UC) and to a lesser extent Crohn's disease. As many as 5% of patients with IBD are co-diagnosed with PSC[34] and approximately 70% of people with PSC have IBD.[12] Of note, the presence of colitis appears to be associated with a greater risk of liver disease progression and bile duct cancer (cholangiocarcinoma) development, although this relationship remains poorly understood.[35] Close monitoring of PSC patients is vital.
Various forms of gallbladder disease such as gallstones and gallbladder polyps are also common in those with PSC.[3] Approximately 25% of people with PSC have gallstones.[3] Ultrasound surveillance of the gallbladder every year is recommended for people with PSC.[3] Any person with PSC who is found to have a mass in the gallbladder should undergo surgical removal of the gallbladder due to the high risk of cholangiocarcinoma.[3] Osteoporosis (hepatic osteodystrophy) and hypothyroidism are also associated with PSC.
Epidemiology
There is a 2-3:1 male-to-female predilection in primary sclerosing cholangitis.[12] PSC can affect men and women at any age, although it is commonly diagnosed in the fourth decade of life, most often in the presence of inflammatory bowel disease (IBD).[11] PSC progresses slowly and is often asymptomatic, so it can be present for years before it is diagnosed and before it causes clinically significant consequences. There is relatively little data on the prevalence and incidence of primary sclerosing cholangitis, with studies in different countries showing annual incidence of 0.068–1.3 per 100,000 people and prevalence 0.22–8.5 per 100,000; given that PSC is closely linked with ulcerative colitis, it is likely that the risk is higher in populations where UC is more common.[36] In the United States, an estimated 29,000 individuals have PSC.[3]
Society and culture
- Chris Klug – professional snowboarder with PSC who had liver transplant
- Chris LeDoux – professional rodeo rider and country musician with PSC who died of cholangiocarcinoma
- Elena Baltacha – British professional tennis player, diagnosed with PSC at age 19 and died five months after being diagnosed with PSC-associated liver cancer (specifically cholangiocarcinoma) at the age of 30.
- Walter Payton – died of complications of PSC.
Research directions
Although there is no curative treatment, several clinical trials are underway that aim to slow progression of this liver disease.[37] Obeticholic acid is being investigated as a possible treatment for PSC due to its antifibrotic effects. Simtuzumab is a monoclonal antibody against the pro-fibrotic enzyme LOXL2 that is being developed as a possible therapy for PSC.[3]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Rawla, P; Samant, H (January 2020). "Primary Sclerosing Cholangitis". PMID 30725866.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 3 4 5 6 7 8 9 10 11 12 "Definition & Facts for Primary Sclerosing Cholangitis | NIDDK". National Institute of Diabetes and Digestive and Kidney Diseases. Archived from the original on 17 January 2021. Retrieved 11 February 2021.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Lazaridis, KN; LaRusso, NF (September 2016). "Primary Sclerosing Cholangitis". New England Journal of Medicine (Review). 375 (12): 1161–70. doi:10.1056/NEJMra1506330. PMC 5553912. PMID 27653566.
- ↑ Williamson, KD; Chapman, RW (June 2015). "Primary sclerosing cholangitis: a clinical update". British Medical Bulletin. 114 (1): 53–64. doi:10.1093/bmb/ldv019. PMID 25981516.
- 1 2 Tabibian JH, Lindor KD (Sep 2014). "Ursodeoxycholic acid in primary sclerosing cholangitis: If withdrawal is bad, then administration is good (right?)". Hepatology. 60 (3): 785–8. doi:10.1002/hep.27180. PMID 24752961.
- ↑ Tabibian JH, Yang JD, Baron TH, Kane SV, Enders FB, Gostout CJ (2016). "Weekend Admission for Acute Cholangitis Does Not Adversely Impact Clinical or Endoscopic Outcomes". Digestive Diseases and Sciences. 61 (1): 53–61. doi:10.1007/s10620-015-3853-z. PMID 26391268. Epub 2015 Sep 21.
- ↑ Tabibian JH, Abu Dayyeh BK, Gores GJ, Levy MJ (2015). "A novel, minimally-invasive technique for management of peristomal varices". Hepatology. 63 (4): 1398–400. doi:10.1002/hep.27925. PMID 26044445.
- 1 2 Tabiban JH, Lindor KD (Feb 2013). "Primary sclerosing cholangitis: a review and update on therapeutic developments". Expert Review of Gastroenterology & Hepatology. 7 (2): 103–14. doi:10.1586/egh.12.80. PMID 23363260.
- ↑ O'Hara SP, Tabibian JH, Splinter PL, LaRusso NF (Mar 2013). "The dynamic biliary epithelia: Molecules, pathways, and disease". Journal of Hepatology. 58 (3): 575–82. doi:10.1016/j.jhep.2012.10.011. PMC 3831345. PMID 23085249.
- 1 2 Charatcharoenwitthaya P, Lindor KD (Feb 2006). "Primary sclerosing cholangitis: diagnosis and management". Current Gastroenterology Reports. 8 (1): 75–82. doi:10.1007/s11894-006-0067-8. PMID 16510038.
- 1 2 Hirschfield, Gideon M; Karlsen, Tom H; Lindor, Keith D; Adams, David H (2013). "Primary sclerosing cholangitis". The Lancet. 382 (9904): 1587–1599. doi:10.1016/s0140-6736(13)60096-3. PMID 23810223.
- 1 2 3 Robbins SL, Kumar V, Cotran RS (2003). "Chapter 16". Robbins basic pathology (7th ed.). Philadelphia: Saunders. pp. 620–1. ISBN 978-0-7216-9274-6.
- ↑ Tabibian JH, O'Hara SP, Lindor KD (2014). "Primary sclerosing cholangitis and the microbiota: current knowledge and perspectives on etiopathogenesis and emerging therapies". Scandinavian Journal of Gastroenterology. 49 (8): 901–8. doi:10.3109/00365521.2014.913189. PMC 4210190. PMID 24990660.
- ↑ Tabibian JH, Varghese C, O'Hara SP, LaRusso NF (2015). "Microbiome-immune interactions and liver disease". Clinical Liver Disease. 5 (4): 83–85. doi:10.1002/cld.453. PMC 5944616. PMID 29755735.
- ↑ Tabibian JH, Varghese C, LaRusso NF, O'Hara SP (2015). "The Enteric Microbiome in Hepatobiliary Health and Disease". Liver International. 36 (4): 480–7. doi:10.1111/liv.13009. PMC 4825184. PMID 26561779.
- ↑ Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, Larusso NF (Jun 2014). "Cholangiocyte senescence via N-Ras activation is a characteristic of primary sclerosing cholangitis". Hepatology. 59 (6): 2263–75. doi:10.1002/hep.26993. PMC 4167827. PMID 24390753.
- ↑ Tabibian JH, Trussoni CE, O'Hara SP, Splinter PL, Heimbach JK, LaRusso NF (2014). "Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis". Laboratory Investigation. 94 (10): 1126–33. doi:10.1038/labinvest.2014.94. PMC 4184949. PMID 25046437.
- ↑ Tabibian JH, Macura SI, O'Hara SP, Fidler JL, Glockner JF, Takahashi N, Lowe VJ, Kemp BJ, Mishra PK, Tietz PS, Splinter PL, Trussoni CE, LaRusso NF (2013). "Micro-computed tomography and nuclear magnetic resonance imaging for noninvasive, live-mouse cholangiography". Laboratory Investigation. 93 (6): 733–43. doi:10.1038/labinvest.2013.52. PMC 3875307. PMID 23588707.
- ↑ Tabibian JH, Enders F, Imam MH, Kolar G, Lindor KD, Talwalkar JA (2014). "Association between serum IgE level and adverse clinical endpoints in primary sclerosing cholangitis" (PDF). Annals of Hepatology. 13 (3): 384–9. doi:10.1016/S1665-2681(19)30869-5. PMC 4215553. PMID 24756015. Archived from the original (PDF) on 2016-09-14. Retrieved 2016-06-24.
- ↑ Tabibian JH, Asham EH, Goldstein L, Han S, Saab S, Tong MJ, Busuttil R, Durazo FA (2009). "Endoscopic Treatment with Multiple Stents for Post-Liver Transplantation Nonanastomotic Biliary Strictures". Gastrointestinal Endoscopy. 69 (7): 1236–1243. doi:10.1016/j.gie.2008.09.057. PMID 19249040.
- ↑ Lazaridis KN, LaRusso NF (2015). "The Cholangiopathies". Mayo Clinic Proceedings. 90 (6): 791–800. doi:10.1016/j.mayocp.2015.03.017. PMC 4533104. PMID 25957621.
- ↑ Trivedi, Palak J.; Corpechot, Christophe; Pares, Albert; Hirschfield, Gideon M. (2016-02-01). "Risk stratification in autoimmune cholestatic liver diseases: Opportunities for clinicians and trialists". Hepatology. 63 (2): 644–659. doi:10.1002/hep.28128. ISSN 1527-3350. PMC 4864755. PMID 26290473.
- ↑ Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, Mooney J, Sargeant C, Braaten J, Bernard T, King D, Miceli E, Schmoll J, Hoskin T, Thapa P, Enders F (Sep 2009). "High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis". Hepatology. 50 (3): 671–3. doi:10.1002/hep.23082. PMC 2758780. PMID 19585548.
- ↑ Chapman, R; Fevery, J; Kalloo, A; Nagorney, DM; Boberg, KM; Shneider, B; Gores, GJ; American Association for the Study of Liver Diseases (February 2010). "Diagnosis and management of primary sclerosing cholangitis". Hepatology. 51 (2): 660–78. doi:10.1002/hep.23294. PMID 20101749.
- ↑ Lindor, KD; Kowdley, KV; Harrison, ME; American College of Gastroenterology (May 2015). "ACG Clinical Guideline: Primary Sclerosing Cholangitis". American Journal of Gastroenterology. 110 (5): 646–59. doi:10.1038/ajg.2015.112. PMID 25869391.
- ↑ European Association for the Study of the Liver. (August 2009). "EASL Clinical Practice Guidelines: management of cholestatic liver diseases". Journal of Hepatology. 51 (2): 237–67. doi:10.1016/j.jhep.2009.04.009. PMID 19501929.
- ↑ Liver, European Association for the Study of the (2009). "EASL Clinical Practice Guidelines: Management of cholestatic liver diseases". Journal of Hepatology. 51 (2): 237–267. doi:10.1016/j.jhep.2009.04.009. PMID 19501929.
- ↑ Tabibian JH, Visrodia KH, Levy MJ, Gostout CJ (2015). "Advanced endoscopic imaging of indeterminate biliary strictures". World Journal of Gastrointestinal Endoscopy. 7 (18): 1268–78. doi:10.4253/wjge.v7.i18.1268. PMC 4673389. PMID 26675379.
- ↑ Boonstra, Kirsten; Weersma, Rinse K.; van Erpecum, Karel J.; Rauws, Erik A.; Spanier, B.W. Marcel; Poen, Alexander C.; van Nieuwkerk, Karin M.; Drenth, Joost P.; Witteman, Ben J. (2013-12-01). "Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis". Hepatology. 58 (6): 2045–2055. doi:10.1002/hep.26565. ISSN 1527-3350. PMID 23775876.
- ↑ Folseraas, T; Boberg, KM (February 2016). "Cancer Risk and Surveillance in Primary Sclerosing Cholangitis". Clinics in Liver Disease. 20 (1): 79–98. doi:10.1016/j.cld.2015.08.014. PMID 26593292.
- ↑ Tsaitas C, Semertzidou A, Sinakos E (April 2014). "Update on inflammatory bowel disease in patients with primary sclerosing cholangitis". World Journal of Hepatology. 6 (4): 178–87. doi:10.4254/wjh.v6.i4.178. PMC 4009473. PMID 24799986.
- ↑ Kummen, M; Schrumpf, E; Boberg, KM (August 2013). "Liver abnormalities in bowel diseases". Best Practice & Research. Clinical Gastroenterology. 27 (4): 531–42. doi:10.1016/j.bpg.2013.06.013. PMID 24090940.
- ↑ Tabibian JH, Lindor KD. Challenges of Cholangiocarcinoma Detection in Patients with Primary Sclerosing Cholangitis. J Analytical Oncology. 2012;1(1):50-55.
- ↑ Olsson R, Danielsson A, Järnerot G, et al. (1991). "Prevalence of primary sclerosing cholangitis in patients with ulcerative colitis". Gastroenterology. 100 (5 Pt 1): 1319–23. doi:10.1016/0016-5085(91)90784-I. PMID 2013375.
- ↑ Boonstra, Kirsten; Weersma, Rinse K.; van Erpecum, Karel J.; Rauws, Erik A.; Spanier, B.W. Marcel; Poen, Alexander C.; van Nieuwkerk, Karin M.; Drenth, Joost P.; Witteman, Ben J. (2013-12-01). "Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis". Hepatology. 58 (6): 2045–2055. doi:10.1002/hep.26565. ISSN 1527-3350. PMID 23775876.
- ↑ Feld JJ, Heathcote EJ (October 2003). "Epidemiology of autoimmune liver disease". Journal of Gastroenterology and Hepatology. 18 (10): 1118–28. doi:10.1046/j.1440-1746.2003.03165.x. PMID 12974897.
- ↑ Trivedi, Palak J.; Hirschfield, Gideon M. (2013-05-01). "Treatment of autoimmune liver disease: current and future therapeutic options". Therapeutic Advances in Chronic Disease. 4 (3): 119–141. doi:10.1177/2040622313478646. ISSN 2040-6223. PMC 3629750. PMID 23634279.
External links
| Classification | |
|---|---|
| External resources |