Short QT syndrome
| Short QT syndrome | |
|---|---|
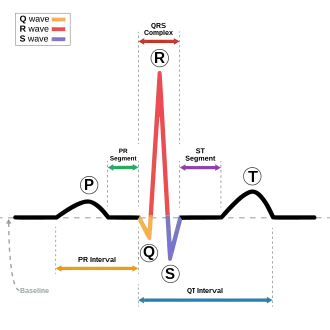 | |
| Schematic representation of normal ECG trace (sinus rhythm), with waves, segments, and intervals labeled | |
| Specialty | Cardiology |
| Symptoms | Passing out, sudden cardiac death[1] |
| Causes | Genetic |
| Diagnostic method | Electrocardiogram (ECG)[1] |
| Treatment | Medication, implantable cardioverter defibrillator (ICD)[1] |
| Medication | Quinidine, Flecainide[1] |
| Prognosis | Risk of sudden cardiac death 0.8% per year[2] |
| Frequency | <300 cases reported worldwide[1] |
Short QT syndrome (SQT) is a very rare genetic disease of the electrical system of the heart, and is associated with an increased risk of abnormal heart rhythms and sudden cardiac death.[1] The syndrome gets its name from a characteristic feature seen on an electrocardiogram (ECG) – a shortening of the QT interval. It is caused by mutations in genes encoding ion channels that shorten the cardiac action potential, and appears to be inherited in an autosomal dominant pattern.[1] The condition is diagnosed using a 12-lead ECG.[2] Short QT syndrome can be treated using an implantable cardioverter-defibrillator or medications including quinidine.[3] Short QT syndrome was first described in 2000,[4] and the first genetic mutation associated with the condition was identified in 2004.[5]
Signs and symptoms
Those affected by short QT syndrome (SQT) have an increased risk of developing abnormal heart rhythms.[3] These abnormal heart rhythms often occur at a young age. They may take relatively benign forms such as atrial fibrillation, leading to symptoms of palpitations, breathlessness, or fatigue.[3] Accordingly, atrial fibrillation presenting in a newborn should raise the suspicion of short QT syndrome.[1] In addition, far more dangerous heart rhythm disturbances such as ventricular fibrillation can also occur in those with short QT syndrome, leading to blackouts or even sudden death.[3] More than a third of those with short QT present with ventricular arrhythmias or sudden cardiac death, while one in five cases are detected during family screening, and one in five cases are found incidentally after an electrocardiogram (ECG) has been recorded for another reason.[1]
If someone with short QT syndrome is examined while their heart is beating in an abnormal rhythm such as atrial fibrillation, this can be detected by feeling their pulse. No abnormal signs will usually be found when examining someone with short QT syndrome while their heart is beating in its normal or sinus rhythm.
Cause
Short QT syndrome is a genetic disorder caused by mutations in genes responsible for producing certain ion channels within heart cells. It appears to be inherited in an autosomal dominant pattern.[1] Some genetic variants cause an increased flow of potassium out of the cell, while others reduce the flow of calcium into the cell.[1] The common effect of all these variants is to shorten the cardiac action potential, reflected on the surface ECG as a shortening of the QT interval. A list of genes in which variants have been associated with short QT syndrome can be found in the table below.
| Type | OMIM | Gene | Notes |
|---|---|---|---|
| SQT1 | 609620 | KCNH2 | Also known as hERG, encodes the potassium channel KV11.1 responsible for the delayed rectifier potassium current IKr [1] |
| SQT2 | 609621 | KCNQ1 | Encodes the potassium channel responsible for the delayed rectifier potassium current IKs [1] |
| SQT3 | 609622 | KCNJ2 | Encodes the potassium channel Kir2.1 responsible for the inward rectifying potassium current IK1 [1] |
| SQT4 | 114205 | CACNA1C | Encodes the alpha subunit of the L-type calcium channel carrying ICa(L) [1] |
| SQT5 | 114204 | CACNA2D1 | Encodes the alpha2/delta subunit of the L-type calcium channel carrying ICa(L) [1] |
| SQT6 | 106195 | SLC4A3 | Encodes a bicarbonate / chloride exchanger [1] |
Mechanism
The overall effect of each of the genetic variants associated with short QT syndrome is to shorten the cardiac action potential, which in turn increases the risk of developing abnormal heart rhythms including atrial fibrillation and ventricular fibrillation.[2] During the normal rhythm of the heart, or sinus rhythm, smooth waves of electrical activity pass regularly through the cardiac muscle. In contrast, during atrial or ventricular fibrillation, waves of electrical activation spiral through the cardiac muscle chaotically in a mass of disorganised, broken wavelets. The consequence of fibrillation is that the chambers of the heart affected by the disorganised electrical activation lose their pumping ability – fibrillation of the cardiac atria in atrial fibrillation leads to an irregular pulse, and fibrillation of the cardiac ventricles in ventricular fibrillation renders the heart unable to pump blood at all.[6]
There are several possible mechanisms by which short action potentials might promote fibrillation. The link between these mechanisms is how the duration of the action potential influences how frequently a heart muscle cell can be excited. A shorter action potential generally allow a heart muscle cell to be excited more frequently – the refractory period is shorter.[6]
The first mechanism, referred to as the dispersion of repolarisation, occurs because the action potential shortening seen in this condition occurs to a greater extent in some layers of the heart wall than in others.[7] This means that at certain points in the cardiac cycle, some layers of the heart wall will have fully repolarised, and are therefore ready to contract again, while other regions are only partially repolarised and therefore are still within their refractory period and not yet able to be re-excited. If a triggering impulse arrives at this critical point in the cardiac cycle, the wavefront of electrical activation will conduct in some regions but block in others, potentially leading to wavebreak and re-entrant arrhythmias.[8]
The second mechanism relates to the increased number of fibrillatory wavelets that can simultaneously exist if the action potential decreases, in a concept known as the arrhythmia wavelength.[6] During fibrillation, the chaotic wavelets rotate, or re-enter, within the muscle of the heart, continually extinguishing and reforming. The volume of tissue in which each wavelet can complete a re-entrant circuit is dependent on the refractory period of the tissue and the speed at which the waves of depolarisation traverse move – the conduction velocity.[6] The product of the conduction velocity and refractory period is known as the wavelength. In tissue with a lower wavelength a wavelet can re-enter within a smaller volume of tissue. A shorter refractory period therefore allows more wavelets to exist within a given volume of tissue, reducing the chance of all wavelets simultaneously extinguishing and terminating the arrhythmia.[6]
Diagnosis
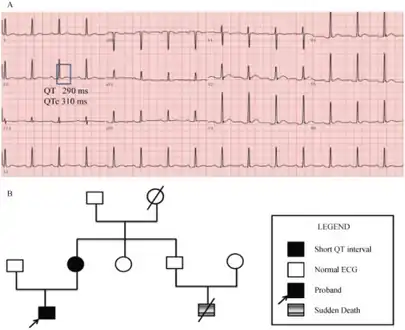 Pedigree of the short QT syndrome family a) ECG of the proband b) family investigation
Pedigree of the short QT syndrome family a) ECG of the proband b) family investigation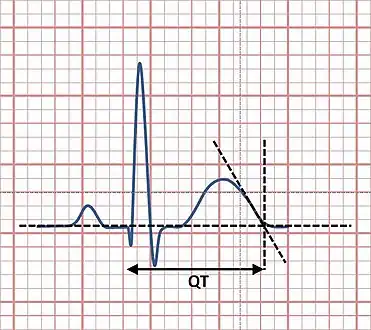 Measurement of the QT interval using the tangent method
Measurement of the QT interval using the tangent method.svg.png.webp) Precordial leads of a 12-lead ECG from a person with short QT syndrome
Precordial leads of a 12-lead ECG from a person with short QT syndrome
Short QT syndrome is diagnosed primarily using an electrocardiogram (ECG), but may also take into account the clinical history, family history, and possibly genetic testing. Whilst a diagnostic scoring system has been proposed that incorporate all of these factors (the Gollob score[9]), it is uncertain whether this score is useful for diagnosis or risk stratification,[10] and the Gollob score has not been universally accepted by international consensus guidelines.[11][12] There continues to be uncertainty regarding the precise QT interval cutoff that is should be used for diagnosis.[12]
12-lead ECG
The mainstay of diagnosis of short QT syndrome is the 12-lead ECG. The precise QT duration used to diagnose the condition remains controversial with consensus guidelines giving cutoffs varying from 330 ms,[11] 340 ms or even 360 ms when other clinical, familial, or genetic factors are present.[11][12] The QT interval normally varies with heart rate, but this variation occurs to a lesser extent in those with short QT syndrome.[1] It is therefore recommended that the QT interval is assessed at heart rates close to 60 beats per minute.[1] Other features that may be seen on the ECG in short QT syndrome include tall, peaked T-waves and PR segment depression.[10]
Other features supporting diagnosis
Other features that support a diagnosis of short QT syndrome include: a history of ventricular fibrillation]or ventricular tachycardia despite an apparently structurally normal heart; a family history of confirmed short QT syndrome; a family history of sudden cardiac death aged <40 years; and identification of a genetic mutation consistent with short QT syndrome.[12][11]
Invasive electrophysiological studies, in which wires are passed into the heart to stimulate and record the heart's electrical impulses, are not currently recommended for diagnosing short QT syndrome or predicting the risk of sudden cardiac death.[11][12]
Treatment
The treatment for short QT syndrome is aimed at preventing abnormal heart rhythms and reducing the risk of sudden cardiac death. It has been difficult to experimentally test potential treatments as the condition is very rare, so the evidence for treatment effectiveness comes largely from consensus opinion.[1] In addition to treating the person identified as having the condition, screening of family members may be recommended.
Implantable cardioverter-defibrillator
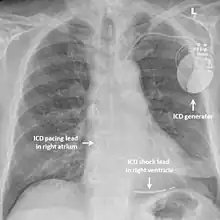
In those with short QT syndrome who have already experienced a life-threatening abnormal heart rhythm such as ventricular fibrillation, an implantable cardioverter-defibrillator (ICD) may be recommended to reduce the chance of sudden death.[3] This device is implanted under the skin and can continually monitor the heart rhythm. If the device detects a dangerous heart rhythm disturbance it can deliver a small electric shock with the aim of restoring a rhythm. Implanting an ICD in someone with short QT syndrome who has not yet experienced a life-threatening arrhythmia is more controversial but may be considered.[1][11]
Medication
Medication aimed at correcting the ECG abnormality – the shortened QT interval – has been tried. Quinidine, a class Ia antiarrhythmic agent, has been shown to partially correct the QT interval and make the heart more resilient to artificially-induced abnormal heart rhythms,[3][13] although it is still uncertain at present whether this translates to a lower risk of sudden death.[1][14] Sotalol, another antiarrhythmic, may prolong the QT in some subtypes of short QT syndrome.[11] Other medications including beta blockers, flecainide, and amiodarone have been tried, but at present there is little evidence to support their use.[1]
Drugs can also be used to treat the less dangerous abnormal heart rhythm that is also associated with short QT – atrial fibrillation. Propafenone, a class 1c antiarrhythmic, may be helpful in those with short QT to prevent atrial fibrillation.[7] Those who develop atrial fibrillation may also require medication to decrease blood clotting in order to reduce the risk of stroke.[15]
Epidemiology
Short QT syndrome is a very rare condition with, as of 2018, fewer than 300 cases described in the medical literature.[1] As a genetic syndrome, those affected are born with the condition. Symptoms can occur in newborns, potentially presenting as sudden infant death syndrome.[2] Males and females are equally likely to be affected, and have a similar risk of sudden cardiac death.[2]
Prognosis
The rarity of short QT syndrome makes calculating prognosis accurately difficult. The risk of sudden cardiac death has been estimated at 0.8% per year,[2] leading to a cumulative risk of sudden cardiac death of 41% by the age of 40.[1] A previous history of cardiac arrest predicts a higher likelihood of further dangerous arrhythmias.[1] Some have suggested that those with the shortest QT intervals may have a higher risk of arrhythmias, but this view has not been supported by all.[1] The findings from invasive electrophysiological studies do not predict an individual with short QT syndrome's risk of cardiac arrest.[1]
History
The first report of short QT syndrome to be published was in 2000, describing a family with short QT intervals on the 12-lead ECG, atrial fibrillation occurring at a young age, and an unrelated patient who had a sudden cardiac death associated with a short QT interval.[2][4] The association between short QT and sudden cardiac death was described in 2003,[16] and the first gene associated with the condition was identified in 2004.[5] Criteria for diagnosing Short QT syndrome were proposed in 2011.[9] Recently the first animal model of short QT syndrome was presented, enabling more in depth analysis of arrhythmia mechanisms.[17]
See also
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 Bjerregaard P (August 2018). "Diagnosis and management of short QT syndrome". Heart Rhythm. 15 (8): 1261–1267. doi:10.1016/j.hrthm.2018.02.034. PMID 29501667. S2CID 4519580.
- 1 2 3 4 5 6 7 Bjerregaard, Preben; Gussak, Ihor (2013), "Short QT Syndrome", Electrical Diseases of the Heart, Springer London, pp. 569–581, doi:10.1007/978-1-4471-4881-4_33, ISBN 9781447148807
- 1 2 3 4 5 6 Schimpf R, Wolpert C, Gaita F, Giustetto C, Borggrefe M (August 2005). "Short QT syndrome". Cardiovascular Research. 67 (3): 357–66. doi:10.1016/j.cardiores.2005.03.026. PMID 15890322.
- 1 2 Gussak, I.; Brugada, P.; Brugada, J.; Wright, R. S.; Kopecky, S. L.; Chaitman, B. R.; Bjerregaard, P. (2000). "Idiopathic short QT interval: a new clinical syndrome?". Cardiology. 94 (2): 99–102. doi:10.1159/000047299. ISSN 0008-6312. PMID 11173780. S2CID 7911999. Archived from the original on 2022-10-23. Retrieved 2022-09-07.
- 1 2 Brugada, Ramon; Hong, Kui; Dumaine, Robert; Cordeiro, Jonathan; Gaita, Fiorenzo; Borggrefe, Martin; Menendez, Teresa M.; Brugada, Josep; Pollevick, Guido D. (2004-01-06). "Sudden death associated with short-QT syndrome linked to mutations in HERG". Circulation. 109 (1): 30–35. doi:10.1161/01.CIR.0000109482.92774.3A. ISSN 1524-4539. PMID 14676148.
- 1 2 3 4 5 Antzelevich, Charles; Burashnikov, Alexander (2013), "Mechanisms of Cardiac Arrhythmia", Electrical Diseases of the Heart, Springer London, pp. 93–128, doi:10.1007/978-1-4471-4881-4_33, ISBN 9781447148807
- 1 2 Enriquez, Andres; Antzelevitch, Charles; Bismah, Verdah; Baranchuk, Adrian (September 2016). "Atrial fibrillation in inherited cardiac channelopathies: From mechanisms to management". Heart Rhythm. 13 (9): 1878–1884. doi:10.1016/j.hrthm.2016.06.008. ISSN 1556-3871. PMID 27291509.
- ↑ Schimpf, Rainer; Wolpert, Christian; Gaita, Fiorenzo; Giustetto, Carla; Borggrefe, Martin (2005-08-15). "Short QT syndrome". Cardiovascular Research. 67 (3): 357–366. doi:10.1016/j.cardiores.2005.03.026. ISSN 0008-6363. PMID 15890322.
- 1 2 Gollob MH, Redpath CJ, Roberts JD (February 2011). "The short QT syndrome: proposed diagnostic criteria". Journal of the American College of Cardiology. 57 (7): 802–12. doi:10.1016/j.jacc.2010.09.048. PMID 21310316.
- 1 2 Rudic, Boris; Schimpf, Rainer; Borggrefe, Martin (August 2014). "Short QT Syndrome - Review of Diagnosis and Treatment". Arrhythmia & Electrophysiology Review. 3 (2): 76–79. doi:10.15420/aer.2014.3.2.76. ISSN 2050-3369. PMC 4711567. PMID 26835070.
- 1 2 3 4 5 6 7 Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. (December 2013). "HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013". Heart Rhythm. 10 (12): 1932–63. doi:10.1016/j.hrthm.2013.05.014. PMID 24011539. Archived from the original on 2021-12-20. Retrieved 2022-09-07.
- 1 2 3 4 5 Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. (November 2015). "2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC)". Europace. 17 (11): 1601–87. doi:10.1093/europace/euv319. PMID 26318695.
- ↑ Gaita F, Giustetto C, Bianchi F, Schimpf R, Haissaguerre M, Calò L, et al. (April 2004). "Short QT syndrome: pharmacological treatment". Journal of the American College of Cardiology. 43 (8): 1494–9. doi:10.1016/j.jacc.2004.02.034. PMID 15093889.
- ↑ Maltret A, Wiener-Vacher S, Denis C, Extramiana F, Morisseau-Durand MP, Fressart V, et al. (February 2014). "Type 2 short QT syndrome and vestibular dysfunction: mirror of the Jervell and Lange-Nielsen syndrome?". International Journal of Cardiology. 171 (2): 291–3. doi:10.1016/j.ijcard.2013.11.078. PMID 24380499.
- ↑ January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC, et al. (December 2014). "2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society". Journal of the American College of Cardiology. 64 (21): e1–76. doi:10.1016/j.jacc.2014.03.022. PMID 24685669.
- ↑ Gaita, Fiorenzo; Giustetto, Carla; Bianchi, Francesca; Wolpert, Christian; Schimpf, Rainer; Riccardi, Riccardo; Grossi, Stefano; Richiardi, Elena; Borggrefe, Martin (2003-08-26). "Short QT Syndrome: a familial cause of sudden death". Circulation. 108 (8): 965–970. doi:10.1161/01.CIR.0000085071.28695.C4. ISSN 1524-4539. PMID 12925462.
- ↑ Odening, Katja E; Bodi, Ilona; Franke, Gerlind; Rieke, Raphaela; Ryan de Medeiros, Anna; Perez-Feliz, Stefanie; Fürniss, Hannah; Mettke, Lea; Michaelides, Konstantin; Lang, Corinna N; Steinfurt, Johannes (2019-03-07). "Transgenic short-QT syndrome 1 rabbits mimic the human disease phenotype with QT/action potential duration shortening in the atria and ventricles and increased ventricular tachycardia/ventricular fibrillation inducibility". European Heart Journal. 40 (10): 842–853. doi:10.1093/eurheartj/ehy761. ISSN 0195-668X. PMID 30496390. Archived from the original on 2022-10-17. Retrieved 2022-09-07.
External links
- "Short QT syndrome". Genetics Home Reference. U.S. National Library of Medicine. Archived from the original on 2022-10-23. Retrieved 2022-09-07.
| Classification |
|---|