Restrictive cardiomyopathy
| Restrictive cardiomyopathy | |
|---|---|
| Other names: Obliterative cardiomyopathy, infiltrative cardiomyopathy, constrictive cardiomyopathy[1] | |
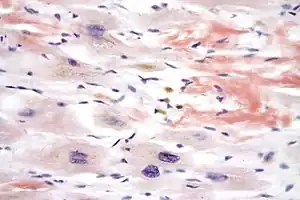 | |
| Micrograph of cardiac amyloidosis, a cause of restrictive cardiomyopathy. Congo red stain. | |
| Specialty | Cardiology |
| Symptoms | Decreased ability to exercise, palpitations, shortness of breath, leg swelling[2][3] |
| Complications | Liver failure, blood clots[2] |
| Causes | Amyloidosis, sarcoidosis, hemochromatosis, Loeffler endocarditis, storage diseases, diabetes, scleroderma, radiation therapy, certain medications, cancer[2] |
| Diagnostic method | Electrocardiogram (ECG), ultrasound of the heart, endomyocardial biopsy[2] |
| Differential diagnosis | Constrictive pericarditis, hypertensive heart disease, hypertrophic cardiomyopathy[2] |
| Treatment | Based on symptoms and the underlying cause[2] |
| Prognosis | Life expectancy 2 to 5 years[2] |
| Frequency | Rare[2] |
Restrictive cardiomyopathy (RCM) is a disease of heart muscle in which the ventricles do not appropriately relax and fill despite relatively normal contraction.[4][3] Symptoms may vary from a decreased ability to exercise, to new onset atrial fibrillation, to heart failure.[2] In some sudden cardiac arrest is the first indication of the disease.[2] Other complications may include liver failure and blood clots.[2]
The most common causes are amyloidosis, sarcoidosis, hemochromatosis, and Loeffler endocarditis.[2] Other causes include storage diseases, diabetes, scleroderma, radiation therapy, certain medications, and cancer.[2] Diagnosis may be supported by electrocardiogram (ECG), ultrasound of the heart, and endomyocardial biopsy.[2]
There is no cure.[2] Treatment involves addressing the underlying cause and measures to improve heart failure.[2] Heart failure may be treated with diuretics such as furosemide, calcium channel blockers, or beta blockers.[2] For sarcoidosis, antiarrhythmics and immunosuppressants are also commonly used.[2] Hemochromatosis may be treated with removal of blood.[2] In certain cases a heart transplant may be an option.[2]
Restrictive cardiomyopathy is relatively rare.[2] It represents about 5% of cardiomyopathy cases with the hypertrophic and dilated types being more common.[2] Life expectancy follow diagnosis is generally 2 to 5 years.[2] Cases of RCM were described in the the 1930s.[5] Cardiomyopathy; however, was not divided into its three main groups until 1961.[1]
Signs and symptoms
Those with RCM will experience decreased exercise tolerance, fatigue, jugular venous distention, peripheral edema, and ascites.[6] Arrhythmias and conduction blocks are common.
Causes
RCM can be caused by genetic or non-genetic factors.[7][8][9] Thus causes can be divide into primary and secondary.[10] It can also be organized into infiltrative, storage diseases, non-infiltrative, and endomyocardial causes.[11] The most common cause of restrictive cardiomyopathy is amyloidosis.[6]
- Infiltrative
- Storage diseases
- Non-infiltrative
- Idiopathic
- Diabetic cardiomyopathy
- Scleroderma
- Myofibrillar myopathies
- Pseudoxanthoma elasticum
- Sarcomeric protein disorders
- Werner's syndrome
- Endomyocardial
- Carcinoid heart disease
- Endomyocardial fibrosis
- Idiopathic
- Hypereosinophilic syndrome
- Chronic eosinophilic leukemia
- Drugs (serotonin, methysergide, ergotamine, mercurial agents, busulfan)
- Endocardial fibroelastosis
- Consequence of cancer or cancer therapy
- Metastatic cancer
- Drugs (anthracyclines)
- Radiation
Mechanism
Rhythmicity and contractility of the heart may be normal, but the stiff walls of the heart chambers (atria and ventricles) keep them from adequately filling, reducing preload and end-diastolic volume. Thus, blood flow is reduced, and blood volume that would normally enter the heart is backed up in the circulatory system. In time, restrictive cardiomyopathy patients develop diastolic dysfunction and eventually heart failure.
Diagnosis
Diagnosis is typically made via echocardiography. Patients will demonstrate normal systolic function, diastolic dysfunction, and a restrictive filling pattern.[11] 2-dimensional and Doppler studies are necessary to distinguish RCM from constrictive pericarditis. Cardiac MRI and transvenous endomyocardial biopsy may also be necessary in some cases.[6][11] Reduced QRS voltage on EKG may be an indicator of amyloidosis-induced restrictive cardiomyopathy.[11]
Untreated hearts with RCM often develop the following characteristics:
- M or W configuration in an invasive hemodynamic pressure tracing of the RA
- Square root sign of part of the invasive hemodynamic pressure tracing Of The LV
- Biatrial enlargement
- Thickened LV walls (with normal chamber size)
- Thickened RV free wall (with normal chamber size)
- Elevated right atrial pressure (>12mmHg),
- Moderate pulmonary hypertension,
- Normal systolic function,
- Poor diastolic function, typically Grade III - IV Diastolic heart failure.
Treatment
Treatment of restrictive cardiomyopathy should focus on management of causative conditions (for example, using corticosteroids if the cause is sarcoidosis), and slowing the progression of cardiomyopathy.[11] Salt-restriction, diuretics, angiotensin-converting enzyme inhibitors, and anticoagulation may be indicated for managing restrictive cardiomyopathy.[12]
Calcium channel blockers are generally contraindicated due to their negative inotropic effect, particularly in cardiomyopathy caused by amyloidosis.[13][14] Digoxin, calcium channel blocking drugs and beta-adrenergic blocking agents provide little benefit, except in the subgroup of restrictive cardiomyopathy with atrial fibrillation.[15] Vasodilators are also typically ineffective because systolic function is usually preserved in cases of RCM.[6]
Heart failure resulting from restrictive cardiomyopathy will usually eventually have to be treated by cardiac transplantation or left ventricular assist device.[12]
Epidemiology
Endomyocardial fibrosis is generally limited to the tropics and sub-saharan Africa.[11] The highest incidence of death caused by cardiac sarcoidosis is found in Japan.[16]
References
- 1 2 Hancock, EW (September 2001). "Differential diagnosis of restrictive cardiomyopathy and constrictive pericarditis". Heart. 86 (3): 343–9. doi:10.1136/heart.86.3.343 (inactive 2020-05-21). PMC 1729880. PMID 11514495.
{{cite journal}}: CS1 maint: DOI inactive as of May 2020 (link) - 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Brown, KN; Pendela, VS; Diaz, RR (January 2020). "Restrictive Cardiomyopathy". PMID 30725919.
{{cite journal}}: Cite journal requires|journal=(help) - 1 2 "Restrictive Cardiomyopathy - Heart and Blood Vessel Disorders". Merck Manuals Consumer Version. Archived from the original on 29 March 2015. Retrieved 10 February 2021.
- ↑ Kouchoukos, Nicholas T.; Blackstone, Eugene H.; Hanley, Frank L.; Kirklin, James K. (2012). Kirklin/Barratt-Boyes Cardiac Surgery E-Book. Elsevier Health Sciences. p. 803. ISBN 978-1-4557-4605-7. Archived from the original on 2021-08-29. Retrieved 2021-02-10.
- ↑ Candell-Riera, J.; Ortega-Alcalde, D. (2012). Nuclear Cardiology in Everyday Practice. Springer Science & Business Media. ISBN 978-94-011-1984-9. Archived from the original on 2021-08-29. Retrieved 2021-02-10.
- 1 2 3 4 Pathophysiology of heart disease : a collaborative project of medical students and faculty. Lilly, Leonard S., Harvard Medical School. (5th ed.). Baltimore, MD: Wolters Kluwer/Lippincott Williams & Wilkins. 2011. ISBN 978-1605477237. OCLC 649701807.
{{cite book}}: CS1 maint: others (link) - ↑ Brodehl, Andreas; Ferrier, Raechel A.; Hamilton, Sara J.; Greenway, Steven C.; Brundler, Marie-Anne; Yu, Weiming; Gibson, William T.; McKinnon, Margaret L.; McGillivray, Barbara (March 2016). "Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy". Human Mutation. 37 (3): 269–279. doi:10.1002/humu.22942. ISSN 1098-1004. PMID 26666891.
- ↑ Brodehl, Andreas; Gaertner-Rommel, Anna; Klauke, Bärbel; Grewe, Simon Andre; Schirmer, Ilona; Peterschröder, Andreas; Faber, Lothar; Vorgerd, Matthias; Gummert, Jan (2017). "The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy". Human Mutation. 38 (8): 947–952. doi:10.1002/humu.23248. ISSN 1098-1004. PMID 28493373.
- ↑ Brodehl, Andreas; Pour Hakimi, Seyed Ahmad; Stanasiuk, Caroline; Ratnavadivel, Sandra; Hendig, Doris; Gaertner, Anna; Gerull, Brenda; Gummert, Jan; Paluszkiewicz, Lech; Milting, Hendrik (2019-11-11). "Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect". Genes. 10 (11): 918. doi:10.3390/genes10110918. ISSN 2073-4425. PMC 6896098. PMID 31718026.
- ↑ Crawford, Michael H. (2003). Current diagnosis & treatment in cardiology. New York: Lange Medical Books/McGraw-Hill. pp. 188. ISBN 978-0-8385-1473-3.
- 1 2 3 4 5 6 Muchtar, Eli; Blauwet, Lori A.; Gertz, Morie A. (2017-09-15). "Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy". Circulation Research. 121 (7): 819–837. doi:10.1161/CIRCRESAHA.117.310982. ISSN 0009-7330. PMID 28912185.
- 1 2 "Restrictive Cardiomyopathy Treatment & Management". 2014-12-18. Archived from the original on 2015-06-10. Retrieved 2015-06-10.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Pollak, A; Falk, R H (1993-08-01). "Left ventricular systolic dysfunction precipitated by verapamil in cardiac amyloidosis". Chest. 104 (2): 618–620. doi:10.1378/chest.104.2.618. ISSN 0012-3692. PMID 8339658.
- ↑ Gertz, Morie A.; Falk, Rodney H.; Skinner, Martha; Cohen, Alan S.; Kyle, Robert A. (1985-06-01). "Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents". American Journal of Cardiology. 55 (13): 1645. doi:10.1016/0002-9149(85)90995-6. ISSN 0002-9149. PMID 4003314.
- ↑ Artz, Gregory; Wynne, Joshua (October 2000). "Restrictive Cardiomyopathy". Current Treatment Options in Cardiovascular Medicine. 2 (5): 431–438. doi:10.1007/s11936-000-0038-6. ISSN 1092-8464. PMID 11096547.
- ↑ Hulten, Edward; Aslam, Saira; Osborne, Michael; Abbasi, Siddique; Bittencourt, Marcio Sommer; Blankstein, Ron (February 2016). "Cardiac sarcoidosis—state of the art review". Cardiovascular Diagnosis and Therapy. 6 (1): 50–63. doi:10.3978/j.issn.2223-3652.2015.12.13. ISSN 2223-3652. PMC 4731586. PMID 26885492.
External links
| Classification | |
|---|---|
| External resources |
- Overview Archived 2011-10-04 at the Wayback Machine at Merck Manual