تليف كيسي
التليف الكيسي هو مرض وراثي صبغي جسمي متنحّ يحدث بسببه عجز مترقٍّ في عمل الغدد خارجية الإفراز، مما يُؤثر على وظائف متعددة في الجسم.[1] فهو يُؤثر بصورة كبيرة على الرئتين، وبنسبة أقل على البنكرياس والكبد والأمعاء، حيث يُؤدي إلى تراكم طبقة سميكة ولزجة من المُخاط على تلك الأعضاء. إنه أحد أكثر الأمراض الرئوية المزمنة انتشارًا لدى الأطفال والشباب عبارة عن اضطراب جيني يُؤدي للوفاة. وفي الغالب يرجع سبب الوفاة إلى الالتهاب الرئوي الذي تُسببه الزائفة والمكورات العنقودية.[2]
| تليف كيسي | |
|---|---|
| Cystic Fibrosis | |
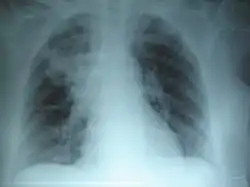 ذات الرئة السفلي الأيمن: يُؤدي التليف الكيسي المُزمن إلى تدمير النسيج البرانشيمي للرئة، مُسببًا في النهاية موت المريض نتيجة لتليف الجهاز التنفسي. ذات الرئة السفلي الأيمن: يُؤدي التليف الكيسي المُزمن إلى تدمير النسيج البرانشيمي للرئة، مُسببًا في النهاية موت المريض نتيجة لتليف الجهاز التنفسي. | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية، وطب الأطفال، وطب الرئة |
| من أنواع | اضطراب صبغي جسدي متنحي ، ومرض رئوي ، ومتلازمة، ومرض |
| المظهر السريري | |
| الأعراض | انسداد معوي، وهشاشة العظام، وقرحة هضمية، وركود صفراوي، والتهاب كبدي، وحصاة صفراوية، وتشمع الكبد، والتهاب القصبات، وذات الرئة، وهمود الرئة، وتوسع القصبات، وتليف رئوي، ووهن عضلي، وضيق النفس، وزرقة، وقصور القلب، وفقد النطاف |
| الإدارة | |
| أدوية | |
يحدث التليف الكيسي بسبب وجود طفرة في المورثة (الجين) الذي يعمل على تشفر بروتين منظم الإيصالية عبر الغشاء في التليف الكيسي (CFTR). يُشارك هذا البروتين في مرور أيون الكلوريد من خلال أغشية الخلايا؛ ويُؤدي وجود قُصور فيه إلى إحداث خلل في إنتاج العرق والعصارات الهضمية والمُخاط، ويتطور المرض في حالة تنحي كلا الأليلين.[3] وقد اكتشف ووصف أكثر من 1500 طفرة لهذا المرض؛ معظم تلك الطفرات عبارة عن عناصر صغيرة محذوفة أو طفرات دقيقة، إذ إن أقل من 1% يرجع سببه إلى طفرات في إعادة ترتيب الصبغيات (الكروموسومات).
يُؤثر التليف الكيسي على العديد من الأعضاء والأجهزة مسببًا إنتاج الغدد الإفرازية لمواد غير طبيعية وسميكة. تعد الأعراض الرئوية السبب الرئيس للإصابة بالمرض والوفاة منه إذ تسببت بموت 95% من المصابين به. ويعود السبب إلى حدوث التهابات متكررة ناجمة عن انسداد الشعب الهوائية بسبب إفراز مُخاط سميك للغاية، ومن الأعضاء الأخرى المُتضررة البنكرياس والخصية عند الذكور.[4][5][6]
وهو أيضًا أحد الأمراض الوراثية الأكثر انتشارًا لدى القوقاز. وعليه فإن نسبة واحدٍ من كل 5000 تقريبًا من أهل تلك البلاد مُصابٍ بذلك المرض يُولد حيًا. ويُقدر ذلك بأن واحدًا من كل 25 فرد من أصل أوروبي يكون حامًلا لأليل متنحٍّ.
يُشير مصطلح التليف الكيسي إلى العمليات التي تتسم بوجود ندوب داخلية أو تليف مع تكوين كيسات داخل البنكرياس. اكتشف ذلك عام 1930، كما يُطلق أيضًا عليه اسم لزوجة المُخاط.
يُطلب من المرضى الذين لديهم نسب عالية من الملح (كلوريد الصوديوم) في العرق بإجراء تشخيص من خلال اختبار فحص العرق. وأيضًا من خلال عمل فحوصات جينية قبل الولادة وبعدها عن طريق إجراء اختبار العرق جيبسون وكوك.
لايوجد حتى الآن علاج شافٍ من ذلك المرض على الرغم من وجود طرق علاجية تعمل على تحسين الأعراض وإطالة متوسط عمر المريض. وفي الحالات الخطيرة ربما أدى تفاقم المرض إلى ضرورة زرع رئة، وفي جميع أنحاء العالم يصل متوسط عمر هؤلاء المرضى إلى 35 عامًا، في حين إن البلدان التي تتبع أنظمة صحية مُتطورة وصلت إلى نتائج أفضل؛ فعلى سبيل المثال في كندا بلغ مُتوسط عمر الفرد المصاب 48 عامًا في 2010.[7][8][9]
الأعراض والعلامات[10]
تتباين أعراض التليف الكيسي وفقًا لعمر الشخص ولمدى تأثيرها على الأعضاء والطرق العلاجية المُستخدمة وأنواع العدوى المُرتبطة به. ويُعيق هذا المرض الجسم بشكل تام، ويظهر أثره على النمو ووظيفتي التنفس والهضم. وتتسم مرحلة ما بعد الولادة بصِغر وزن المولود وبانسداد معوي ناجم عن سُمك البراز وغَلاظتِه، وتظهر بعض الأعراض في مرحلة مُتقدمة؛ فمنها ما يظهر أثناء الطفولة ومنها ما يظهر في بداية البلوغ. وجميع الأعراض تُؤدي إلى تأخر النمو وظهور الأمراض الرئوية وزيادة المشاكل التي يُسببها سوء امتصاص الفيتامينات والعناصر الغذائية في الجهاز الهضمي.[11]
يشخص التليف الكيسي عند معظم الأطفال قبل السنة الأولى من العمر، وذلك عند ظهور نتائج تأثير لزوجة المُخاط على الرئتين والبنكرياس. ففي الجهاز التنفسي تُصبح هذه الإفرازات بمنزلة أرض خصبة للعديد من البكتيريا المسؤولة عن العدوى المُزمنة فتُسبب تلفًا تدريجيًا ومُستمرًا للنسيج البارينشيمي بالخلايا الرئوية، وتزداد الحالة التنفسية سوءًا حيث إن المصابين يُعانون من ارتفاع ضغط الدم الرئوي. وفي البنكرياس -من ناحية أخرى- يُعيق المُخاط مرور الإنزيمات التي تفرزها الغُدة، ويمنع وصولها إلى الأمعاء لهضم وامتصاص الطعام.[11]
أمراض الرئة والجيوب الأنفية

تنتج أمراض الرئة من انسداد الشعب الهوائية الصغيرة بالمُخاط السميك مُسببة بذلك التليف الكيسي. ويُسبب الالتهاب والعدوى تلف بالرئتين، كما يُحدث تغَيُرات هيكلية تُؤدي فيما بعد إلى ظهور عدة أعراض مُتنوعة. ففي المراحل الأولى، عادًة ما يُعاني المريض من سُعال مُتواصل وإفراز كميات كبيرة من المُخاط وصعوبة القدرة على التنفس. وتحدث العديد من هذه الأعراض بشكل سريع عند نمو أنواع معينة من البكتيريا، (وبخاصة الزائفة الزنجارية) التي تعيش عادة في المُخاط السميك، مُسببة بذلك ذات الرئة. وفي مراحل مُتقدمة من التليف الكيسي، تحدث تغيرات في بنية الرئة تُؤدي، فيما بعد، إلى حدوث صُعوبات تنفسية مُزمنة.
هناك العديد من الأعراض الأخرى منها: نفث الدم أو بصق الدم وتوسع حاد ومزمن في الشعب الهوائية أو القصبيات (القصبات) وارتفاع ضغط دم الرئة وحدوث قصور القلب والشعور بعدم الحصول على كمية كافية من الأكسجين أو ضيق التنفس أو الفشل التنفسي أو انخماص الرئة الذي قد يتطلب اللجوء للتنفس الاصطناعي.[12] وبالإضافة إلى تلك العدوى البكتيرية الأكثر شيوعًا، تتطور أنواع جديدة من أمراض الجهاز التنفسي لدى الأشخاص المصابين بالتليف الكيسي بسهولة كبيرة. ومن بين ذلك نجد داء الرشاشيات القصبي الرئوي التحسسي، وهو عبارة عن شدة تحسس الجهاز المناعي لنوع من الفطريات (عفن) ينتمي لجنس الرشاشيات (الرشاشية الدخناء)، مما يُؤدي إلى تزايد المشاكل التنفسية. وأيضًا نجد الإصابة بعدوى مركب المُتفطرات الطيرية، وهي عبارة عن مجموعة من البكتيريا الشعاوية التي تتصل بنوع المُتَفَطِّرة السُلِّية والتي يمكن أن تُلحق أضرار كبيرة بالرئة، وهي لا تستجيب عادةً للمضادات الحيوية التقليدية.
وتُؤدي أيضا لزوجة وسُمك المُخاط في الجيوب الأنفية، إلى انسداد الثقوب التي عادة ما يتم الصرف من خلالها، الأمر الذي يجعل من تراكم تلك الإفرازات أرض خصبة لمسببات الأمراض الآنف ذكرها. وفي هذه الحالات، قد يُؤدي ذلك إلى ظهور آلام بالوجه وحمى ورشح شديد وصداع. ويُلاحظ غالبًا على الأشخاص المصابين بالتليف الكيسي نمو مفرط في النسيج الأنفي (بوليب)، ويظهر ذلك نتاج للالتهاب الذي يحدث إثر الإصابة بعدوى الجيوب الأنفية المزمنة. ويمكن أن تزيد هذة البوليبات بسبب انسداد الشعب الهوائية العليا، وتُضاعف من صعوبات التنفس.[13][14]
أمراض الجهاز الهضمي والكبد والبنكرياس
قبل انتشار فحوصات ما قبل الولادة وما بعدها للكشف عن مرض التليف الكيسي، كان يتم الكشف عنه عادًة في حالة عدم قدرة الطفل المولود على طرد أول براز له (العِقْيُ)، الذي يمكنه أن يسد الأمعاء ويُؤدي لحدوث اضطرابات خطيرة. وتُسمى هذه الحالة بالعلوص العِقْيُ وتٌصيب 10% من المواليد المصابين بالتليف الكيسي.[15] وتم مؤخرًا تحديد المُتغيرات الوراثية في الجينات التي لها علاقة بنقل الأيونات في الأمعاء الدقيقة التي تؤدي إلى تطور العلوص العِقْيُ.[16] ومن الشائع أيضًا، ارتباط وجود التليف الكيسي مع بروز الأغشية المُستقيمة الداخلية، تدلي المستقيم، ويرجع ذلك إلى زيادة حجم البراز وسوء التغذية وارتفاع الضغط داخل المعدة بسبب السُعال المُزمن.[17]
يرتبط المُخاط اللزج المُتواجد على الرئة بالإفرازات السميكة الخاصة بالبنكرياس، العضو المسئول عن توفير العُصارات الهاضمة التي تساعد في التحلل الكيميائي للطعام. وتمنع هذه الإفرازات حركة الإنزيمات الهضمية إلى الأمعاء، وتنتج بذلك أضرار جَسيمة لا يُمكن إصلاحها في البنكرياس، وغالبًا ما يُصاحب ذلك التهاب شديد.[18] ويُؤدي نقص الإنزيمات الهاضمة إلى إعاقة امتصاص العناصر الغذائية ومع إفراز تلك المواد لاحقًا في البراز يحدث ما هو معروف بسوء الامتصاص الذي يُؤدي إلى نقص التغذية وتأخر النمو، ويرجع كليهما إلى قلة الاستفادة الحيوية من السعرات الحرارية. لذلك، فإن الأشخاص المُصابين بالتليف الكيسي، على وجه الخصوص، يُعانون من مشاكل في امتصاص فيتامين ألف وفيتامين دي، وإي، وك. وإلى جانب إصابة البنكرياس، عادًة ما يُعاني هؤلاء المرضى من ارتجاع مريئي حاد وجفاف الحلق وانسداد الأمعاء بسبب الانغلاف المعوي والإمساك.[19] وتَطَورت أعراض الانسداد المعوي عند المرضى كبار السن، وذلك بسبب لزوجة البراز.[20]
يُمكن أن تؤدي هذه الإفرازات أيضًا إلى حدوث مُشكلات في الكبد. فالعُصارة الصفراوية التي تُفرزها الأحشاء لتسهيل الهضم، من المُمكن أن تُؤدي إلى انسداد القنوات الصفراوية وتُلحق الضرر بالأنسجة المُجاورة لها. ويُؤدي هذا في نهاية الأمر إلى حدوث تليف كبدي. وفي تلك الحالة، تتعرض كافة الأعضاء لخطر من الدرجة الأولى من قبيل: التدخل في إبطال مفعول الذيفان، وفي تركيب بعض البروتينات المُهمة، على سبيل المثال عوامل التخثر المسؤلة عن تجلط الدم.[21]
أمراض الغدد الصماء والنمو

يحتوي البنكرياس على جزر لانغرهانس المسئولة عن إفراز الإنسولين، وهو هرمون يُساعد على تنظيم مستويات الجلوكوز في الدم. وقد يُؤدي حدوث أي تلف في البنكرياس إلى فقدان خلايا جزر لانغرهانس مما يُسبب داء السكري.[22] ومن ناحية أخرى، يُساعد فيتامين دي في تنظيم نسبة الكالسيوم والفوسفور في الجسم. ويُؤدي نقص فيتامين دي، الذي يرجع لضعف التغذية، إلى هشاشة العظام مما يزيد من خطر الإصابة بكسور.[23] علاوة على ذلك، فإن الأشخاص المُصابين بالتليف الكيسي عادًة ما يظهر في أيديهم وأرجلهم تشوهات تُعرف بتعجر الأصابع، التي تُعود أسبابها إلى ذلك المرض المُزمن وأيضًا إلى نقص التأكسج في عظام المريض.
من السمات المميزة أيضًا لهذا المرض تأخر النمو. فبشكل عام، يُلاحظ على الأطفال المُصابون بهذا المرض نقص حاد في الوزن مُقارنة بأقرانهم. وفي كثير من الأحيان، يتم التشخيص بشكل أكثر دقة بعد أن يتم التحقق من أسباب هذه الظاهرة. وأسباب تأخر النمو متعددة، فهي تشمل على عدوى الرئة المُزمنة وسوء امتصاص المواد الغذائية في الجهاز الهضمي وزيادة الأيض المُتعلق بتلك الحالة.
يُمكن تشخيص التليف الكيسي عن طريق فحص الأطفال حديثي الولادة وإجراء اختبار لقياس نسبة التعرق بالكهرباء أو الاختبارات الجينية. ففي عام 2006، في الولايات المتحدة الأمريكية، تم فحص 10% من المواليد بعد الولادة مُباشرة، وذلك في إطار برامج فحص المواليد الجدد، والتي من شأنها أن تُحدد مدى ارتفاع إنزيم التربسن. ومع ذلك، فإن معظم البلدان لا يجري فيها مثل هذه الفحوصات بشكل روتيني. ولهذا السبب، فإن الأشخاص المُصابين لا يتلقون التشخيص المُناسب إلا بعد ظهور أعراض المرض عليهم. فالفحص التشخيصي الأكثر شيوعًا واستخدامًا لهذا المرض هو اختبار العرق، كما وصفه دكتور لويس إي جيبسون ودكتور روبرت كوك عام 1959،[24] عبر استخدام رحلان كهربائي كمي، إرحال أيوني، جنبًا إلى جنب مع عقار محفًز للتعرق، بيلوكاربين. وتتم هذه العملية عن طريق تطبيق هذه المادة، التي تحمل شُحنة مُوجبة على قطب كهربائي مُوجب (+) في اتصال مع الجلد. وبعد تمرير التيار الكهربائي، يُهاجر العقار من الغشاء عبر قطب آخر يحمل شُحنة سالبة (-)، بحيث يتم وضع مسافة مُحددة حتى مرورها عبر البشرة، وينتج عن ذلك تحفيز الغُدد العرقية مسببًة إفراز نسبة مُحددة من العرق. بعد ذلك، يتم تجميع عينات العرق على ورق ترشيح أو في أنبوب شعري ومن ثَم يتم تحليلها وتحديد مدى تركيز الصوديوم والكلوريد. فالأشخاص المُصابون بالتليف الكيسي لديهم نسب أعلى من هذه الأيونات في العرق. وبمجرد أن تُظهرالفحوصات نتائج إيجابية لاختبار العرق، يتم التشخيص بشكل أكثر تفصيًلا ودقة، وذلك من خلال تحديد الطفرات في الجين الذي يعمل على تشفر بروتين منظم الإيصالية عبر الغشاء في التليف الكيسي.(CFTR).[25]
هناك العديد من الاختبارات لتحديد المُضاعفات المُحتملة ومُراقبة تطور المرض. فالصور التي حصلت عليها الأشعة السينية والمقطعية تُسهل الكشف عن علامات الإصابة أو عدوى الرئتين. ويتم زرع القشع باستخدام الميكروسكوب، فهو اختبار يُساعد في الكشف عن نوع البكتيريا التي تُصيب الرئة أو الشُعب الهوائية، كما يساعد في اختيار المضادات الحيوية الأكثر فعالية للمرض. فتقيس اختبارات قياس الوظائف التنفسية سعات الرئة وحجمها وسرعة الرئتين في تعبئة الهواء وتدفقه. ومن خلال هذة الاختبارات، يُمكن تحديد ما إذا كان العلاج بالمضادات الحيوية مُلائم أو عمل تقييم لاستجابة المريض لهذه المضادات. فاختبارات تحليل الدم تُساعد في التعرف على مشاكل في الكبد ونقص الفيتامينات وتكشف عن ظهور مرض السكري. يقوما الجهازان ديسكا أو ديسك بقياس امتصاص الأشعة السينية ثنائي البواعث لصحة العظام، والتي تُعتبر كدليل لوجود مرض هشاشة العظام. وفي النهاية، فإن تحديد نسبة الإيلاستاز في البراز، يُساعد في الكشف عن قصور الإنزيمات الهضمية.
الفسيولوجية المرضية
يرتبط البروتين المركب من الجين CFTR بالغشاء الخلوي في كُلا من الغُدد العرقية والرئة والبنكرياس إلى جانب أعضاء أخرى تتضرر بذلك أيضًا. يُغطي البروتين هذا الغشاء ويعمل كقناة أيونية لربط الجزء الداخلي من الخلية، السيتوبلازم، مع السائل خارج الخلية. هذة القناة هي المسؤلة بشكل رئيسي عن التحكم في مرور الكلوريد من وإلى الخلية. وفي حالة حدوث خلل في البروتين CFTR، يتم عرقلة هذة الحركة ومن ثم ترسب الكلوريد خارج الخلية. وبسبب حمل الكلوريد لشُحنة كهربائية سلبية، فإن الأيونات التي تحمل شُحنات مُوجبة لا يمكنها أيضًا أن تمر عبر الغشاء السيتوبلازمي، ويرجع ذلك إلى الشُحنة الكهربائية التي تبذلها أيونات الكلوريد. فالصوديوم هو من أكثر الأيونات الموجودة خارج الخلية شيوعًا، وبالجمع بين الكلوريد والصوديوم، ينتج كلوريد الصوديوم. وفي حالة الإصابة بالتليف الكيسي، يفقد المريض كميات كبيرة من كلوريد الصوديوم في العرق. ويُعتبر الملح المفقود هو السبب الرئيسي لشرح الفائدة التشخيصية لاختبار العرق.[12]
فالتقنية التي من خلالها يُؤدي الخلل الخلوي لظهور الأعراض السريرية، الآنف ذكرها، ليست معروفة بدقة. وتُشير إحدى النظريات إلى أن فشل البروتين مُنظم الإيصالية عبر الغشاء في التليف الكيسي في نقل الكلوريد، وبالتحديد تراكم كمية كبيرة من المُخاط على الرئتين، أدى إلى خلق بيئة غنية بالمواد الغذائية مُلائمة للبكتيريا تستطيع من خلالها الهرب من الجهاز المناعي. ومن المفترض أيضًا أن هذا الخلل، الذي يحدث في البروتين، يُؤدي إلى زيادة مُتناقضة في اقتناص الكلوريد والصوديوم، مما يُحفز إعادة امتصاص المياه، والتي ينتج عنها نقص الماء بالمُخاط وترسبه بشكل جاف وسميك. وتركز نظريات أخرى على ظاهرة حركة الكلوريد خارج الخلية، التي من شأنها أن تُؤدي أيضًا إلى جفاف المُخاط والإفرازات البنكرياسية والصفراوية. وتتفق هذه النظريات، بشكل عام، في إرجاع سبب الإضرابات الكبيرة التي تُعرقل القنوات الناقلة الدقيقة إلى الإفرازات اللزجة السميكة التي تترسب على العديد من الأعضاء المُتضررة. كل ذلك يُؤدي إلى حدوث عدوى مُزمنة ويعزز إعادة هيكلة الرئة، بالإضافة إلى حدوث أضرار بنكرياسية، بسبب تكتل الإنزيمات الهضمية وانسداد الأمعاء بكميات كبيرة من البراز.[12]
دور العدوى المزمنة في مرض الرئة
تستعمر البكتيريا في سن مبكر كلتا الرئتين عند الأشخاص المُصابين بالتليف الكيسي. فتنتشر هذه الكائنات الدقيقة لدى الأشخاص المُصابين وتتكاثر بشكل أكبر في المُخاط المُتراكم في الشعب الهوائية الضيقة. ويحفز المُخاط اللزج هذه الكائنات على التكاثر في الأغشية الحيوية، مما يُؤدي إلى صعوبة اختراقها بواسطة المضادات الحيوية وخلايا جهاز المناعة. وفي الوقت نفسه، تستجيب الرئتان للتلف المُستمر، الذي تُسببه الإفرازات السميكة والالتهابات المزمنة، التي تتكون تدريجيًا في الشعب الهوائية السفلية، حيث توسع القصبات، مما يُصعب القضاء على هذه العدوى.[26]

مع مرور الوقت، تتغير نوع البكتيريا التي تصيب المرضى وتتغير أيضًا خصائصها. ففي المرحلة الأولي، تُستعمر الرئتان من قِبل أنواع مُحددة من البكتيريا مثل المكورات العنقودية الذهبية والمستديمة النزلية. وتنتشر فيما بعد الزائفة الزنجارية، وأحيانًا تتكون البيركهولدرية البصلية من عدة أنواع مُختلفة من البيركهولدرية. وعند انتشار هذه البكتيريا داخل الشعب الهوائية، سُرعان ما تتكيف مع البيئة وتطور مقاومتها للمضادات الحيوية. ويُمكن أن تُطور الزائفة بعض من خصائصها المُميزة مما يُؤدي إلى تكوين مُستعمرات كبيرة، وتُعرف هذه السلالات باسم الزائفة المُخاطية، وهي نادرًا ما تظهر عند الأشخاص الأصحاء.[26]
تنتشر العدوى بشكل كبير بين الأشخاص المُصابين بالتليف الكيسي.[27] فقد كان من الأمور المعتادة في السابق أن يتشارك الأشخاص المُخيمات الصيفية والأنشطة الترفيهية معًا.[28][29] وكانت المستشفيات تحتجز المرضى المُصابين بالتليف الكيسي في غُرف مشتركة، بالإضافة إلى عدم تعقيم المُعدات الطبية، (على سبيل المثال: الرذاذات)[30] عند استخدامها بشكل متكرر[31] مما أدى إلى انتقال سلالات بيكتيرية خطيرة جدًا بين المرضى. ولكن في الوقت الحالي، تقوم منشآت الرعاية الصحية بعزل هؤلاء المرضى عن بعضهم البعض؛ علاوة على إنه يجب على الأشخاص المسئولة عن رعايتهم ارتداء معاطف وقفازات للحد من انتشار السلالات البكتيرية الخبيثة.[32] وفي كثير من الأحيان، يتلقى الأشخاص المصابون بتلك البكتيريا الخطيرة الرعاية في أيام مُتباينة ومبانٍ مُختلفة عن غيرهم. وعلاوًة على العدوى البكتيرية، فإن مرضى التليف الكيسي هم عُرضة أكثر من غيرهم للاستعمارالفطري، وذلك بسبب قدرة بعض الفطريات على استعمار الجهاز التنفسي السفلي، ومن ناحية أخرى، تُطَور بعض أنواع البكتيريا من نظام مناعتها وتُخرج أجيالاً تُقاوم بعض أنواع المضادات الحيوية.[33] وتنمو فطريات الرشاشية الدخناء والمبيضة البيضاء بشكل كبير، بحيث يُؤدي هذا الاستعمار الفطري إلى ارتفاع معدل الاستجابة الالتهابية أمام الفطريات.[34] وفي الوقت الحاضر، ليس معروفًا بشكل واضح دور الفطريات في الإصابة بالتليف الكيسي، فهي لا تعتبر المُسببة للمرض إلا في حالات داء الرشاشيات الغازية وداء الرشاشيات القصبي الرئوي التحسسي.
وراثية المرض

التليف الكيسي هو عبارة عن مرض صبغي جسمي متنحي. فهو يحدث نتيجة طفرة في حمض أميني، وهي فقدان فينيل ألانين في الموضع 508، مُسببًا حدوث خلل في البروتين الحامل، وبالتالي تتوطن في الغشاء الخلوي للبروتين CFTR. وتم وصف هذا البروتين بأنه مكون من أكتر من 1800 طفرة،[35] أغلبها عبارة عن حذوفات صغيرة، وعلى الرغم من ذلك، فهي تُحدت تأثيرات مختلفة، كتغيرات في إطار القراءة وتغيرات في الأحماض الأمينية وتغيرات في الربط الجيني.
يقع الجين CFTR على الذراع الطويل من الكروموسوم 7 في الموضع 7q31.2 مُحتلًا بذلك 1800 زوج من أسس القواعد النيتروجينية؛ وبشكل أكثر دقة، من الزوج القاعدي الصبغي 252 907 116 إلى 950 095 117. فهو جين كبير الحجم، حيث يمتلك 250 كيلو قاعدة تحتوي على 27 إكسون. وقد تم تحديد موقعه وتسلسله عن طريق رسم الخرائط الوراثية.
يعمل هذا الجين على تشفير تركيب القناة الأيونية، التي تحتوى على 1480 حمض أميني، فهو البروتين الذي يقوم بنقل أيونات الكلوريد عبر الخلايا الظهارية، ويتحكم أيضًا في تنظيم الناقلات الأخرى. ويختفي هذا الجين أو يظهر بنسب أقل بكثير من المعدل الطبيعي عند الأشخاص المُصابين بالتليف الكيسي. فيتغير اختراق المرض للأشخاص تبعًا للأليل الذي يعتمد بدوره على بيئة وجينوم الشخص المصاب.
البيولوجيا الجزئية

هي عبارة عن عدة آليات، من خلالها تُحدِث الطفرات مُشكلات في بروتين منظم الإيصالية عبر الغشاء في التليف الكيسي. وبشكل خاص، فإن الطفرة ΔF508 تُنشأ بروتين لا ينطوي بشكل طبيعي، بحيث تحلله الخلية في نهاية الأمر. وتحدث العديد من الطفرات المُشتركة بين يهود أشكناز نتيجة لتركيب البروتينات في وقت قصير للغاية. ويرجع ذلك بسبب الإنهاء المُبكر للتخليق الحيوي للبروتين. وهناك طفرات أخرى أقل انتشارًا تُنتج بروتينات لا تستخدم الطاقة بشكل صحيح، حيث أن تلك الطفرات لا تسمح بمرور الكلور عبر الغشاء على النحو المناسب، أو أنها تتحلل بمعدل أسرع من الطبيعي. فحدوث خلل في نقل الكلور يُؤدي إلى عدم طرد الخلايا للماء خارجًا، وبالتالي زيادة لزوجة المُخاط. لذلك فإن بعض الطفرات يُمكن أن تؤدي إلى انخفاض إنتاج نسخ من بروتين (CFTR).[12]
من الناحية الهيكلية، فإن الجين CFTR ينتمي إلى ما يُعرف بفصيلة الناقلات ABC، بالإنجليزية ATP: أدينوسين ثلاثي الفوسفات، ويأتي بمعنى عُلَيْبات أو كاسيتات.[12] ويتكون الهيكل الثالت من البروتين المُشفر بواسطة الجين من مجالين قادرين على تحليل الأدينوسين ثلاثي الفوسفات، الذي يسمح للبروتين باستخدام طاقة في شكل ATP. وعلى نحو مماثل، فإن زوج آخر من هذه المجالات، يتألف كل واحد منهم من ستة من لولب ألفا، الذي يسمح بمرور البروتين من خلال غشاء الخلية. ويتم تحديد التنشيط بفضل نتيجة عملية الفسفرة في موقع ربط منظم، وخاصة من خلال بروتين كيناز A، رقم التصنيف الإنزيمي 2.7.11.11 للـ PKA، المعروفة سابقًا بـ cAPK أو بروتين الكيناز المُعتمد على الأدينوسين أحادي الفسفات الحلقي.[12] ويتحد حمض الكربوكسيلي النهائي (-C) للبروتين مع الهيكل الخلوي عن طريق التفاعل مع نطاقات بروتينية PDZ.[36]
المضاعفات
يُعاني المصابون بالتليف الكيسي من سوء التغذية، لأن المادة المُخاطية تسد قناة البنكرياس مما يمنع الانسياب الطبيعي للخمائر، الإنزيمات، الهاضمة التي تعمل على تفتيت الطعام داخل الأمعاء. كما تسد منافذ الهواء في الرئتين وتُصبحان عُرضة للعدوى السريعة.[37]
إن النمو الضعيف عند الأطفال يظهر كعدم قدرة على زيادة في الوزن أو الطول مُقارنةً بالأطفال الآخرين في نفس السن. كما تتعدد الأسباب العائدة إلى الفشل في النمو وتشمل: الالتهاب المزمن في الرئتين، سوء امتصاص الجهاز الهضمي للمواد الغذائية وزيادة الحاجة إلى الوحدات الحرارية بسبب المرض المزمن.
التشخيص
تشخيص تقليدي (أساليب غير جزيئية)
يُوجد سلسلة من الفحوصات يتم إجرائها بشكل شائع لتحديد الخلل في عمليات الأيض المرتبطة بالتليف الكيسي، خاصة الكلور. ووُجد من بينهم:
- اختبار العرق: حيث يتم فحص البيلوكاربين عن طريق الإرحال الأيوني لتحفيز إنتاج العرق وتُقاس بنفس الطريقة نسبة تركيز الأملاح.[11]
- اختبار فرق الجهد الكهربائي الأنفي.
- اختبار مولِّد التربسين المتفاعل مناعيًا: وهو اختبار يقيس مدي تركيز الإنزيمات البنكرياسية في الدم.
تشخيص جزيئي
التشخيص الجزيئي للمرض مُعقد للغاية، ففي نوفمبر/ تشرين الثاني من عام 2010، تم اكتشاف 1824 طفرة،[38] مُعظمها عُبارة عن حذوفات صغيرة دقيقة. ويتم تصنيف هذه الطفرات وفقًا لتأثيرها على الجين والخصائص الظاهرية للمرض. وبسبب تنوع الطفرات واختلاف السكان عن بعضهم البعض تعين على الدراسات والاختبارات التشخيصية أن تُوجه النظر إلى هذا الجانب. علمًا بأن الطفرة الأكثر شيوعًا بين معظم السكان الحذف 508F. ويجب الوضع في الاعتبار أن الأليلات تختلف كثيرًا باختلاف الأشخاص، لذلك يجب ضبط وتكييف الفُحوصات للكشف عن المُتغيرات الأكثر شيوعًا بين السكان المُجرٍ عليهم الدراسة.
في الوقت الراهن، يُجرى فحص جيني لجميع المواليد عن طريق النظر في التسلسل الجيني لبروتين CFTR، لمعرفة ما إذا كان الطفل حامًلا للمرض وبالتالي يُمكن معالجته. فحينما يبدأ الكشف عن المرض وعلاجه مبكرًا، تزيد فرصة المريض في أن يعيش حياة صحية أطول.
- التشخيص الجزيئي غير المُباشر: يتم إجراءه من خلال تحليل الارتباط الجيني، ويتم ذلك عن طريق:
- تعدد أطوال جزء الحصرRFLP : طريقة قديمة تقوم على الحصر، ولكنها غير مُستخدمة حاليًا.
- عوامل تحديد جيني ساتلي مصغّر SSR/ STR (الساتلايتس المُصغرة:Microsatélite): هي الطريقة المُتبعة حاليًا. ولكن يتم تطبيقها داخل حدود شجرة العائلة للوصول لنتائج جيدة، لإن مناطق الساتلايتس المُصغرة تختلف اختلافًا كبيرًا من شخص لآخر.
- عوامل تعدد أشكال النوكليوتيدات المفردة SNP: سيتم استخدامها في المستقبل القريب، لأنها تتميز بأن نتائجها قابلة للتطبيق على أشخاص ليس بينهم أية صلة.
- التشخيص الجزيئي المُباشر: من الممكن ترتيب التسلسل الجيني لبروتين CFTR، ولكن في الوقت الراهن الكشف عن الطفرات هو الغاية، ويتم تنفذ ذلك وفق استراتيجيتين:
- تَتَبُع الطفرات: وذلك عن طريق تقنيات الكشف عن الطفرات ولكن بدون التعرف عليها. فمن الأشياء الأكثر استخدامًا في هذة العملية هي الفصل الكهربائي الهلامي التدريجي، كما هو الحال في بعض مُتغيرات تفاعل البوليميراز المتسلسل PCR. ومثال على هذا، اكتشاف الطفرة 508F. وتم تنفيذ هذا الاكتشاف بفضل إنزيم الحصر الذي يقطع الأليل الصحي، كما يسمح لأن تُصبح نتائج عملية التفاعل البوليميراز المتسلسل PCR بعد التضخيم أصغر من الأليل الصحي الذي تظهر به الطفرة، وهكذا يظهرالفرق.
- التعرف على الطفرات وتحديدها: هي عبارة عن تقنيات قائمة على أساس تهجين مُحدد مع أليل متغير. على سبيل المثال: مجموعات ASO وOLA، فحص ربط النوكليوتيد، أو يتم استخدام الفحص التقليدي وصمة ساذرون.
- أليل محدد النوكليوتيد ASO وصمة البقع dot blot: تُستخدم للكشف عن طفرات مُحددة سبق ذكرها. بحيث تحتفظ بتضخم الحمض النووي لدراسته. وفيما بعد، يتم تحديده في الغشاء، بدون العلاج أو الفصل الكهربائي الأنف ذكره. وبعد ذلك، يتم تهجينه مع نيوكليوتيدات مُحددة للأليل الذي يحمل 20 زوج من القواعد التي تم توضيحها للكشف عنه سابقًا. فهو عبارة عن آلية بسيطة ورخيصة وسريعة وأمنة في حالة ما إذا تمت دراسة جين واحد وعدد قليل من الطفرات، كما هو الحال في الكشف عن التليف الكيسي. وتكون مُكلفة إذا لزم إعادة عملية التهجين مرات عديدة. ولتلك الحالات، يتم استخدام أليل محدد النوكليوتيد وصمة البقع العكسية التي يتم فيها تحديد الغشاء للأليلات المُختلفة المُهجنة مع الناتج الجيني المُتضخم السابق ذكره.
- OLA فحص ربط النوكليوتيد: هي الطريقة الأكثر استخدامًا في الوقت الراهن لدراسة التليف الكيسي، فهناك خطوتان هامتان ألا وهما: الخطوة الأولى تتكون من البقايا الجينية لتفاعل البوليميراز المتسلسل متعدد الإرسال PCR multiplex. والخطوة الثانية ربط إنزيم حراري مع أليلات ذات نيوكليوتيدات مُحددة. ويتم تنفيذ هذة الخطوات الأولى في نفس أنبوب التفاعل. وبعد ذلك، يتم إجراء دراسة للبقايا بواسطة الرحلان الكهربائي الشعري. فمزايا هذة العملية هي الأتمتة، بالرغم من أنها مُكلفة لحاجتها إلى التسلسل الآلي.
تشخيص ما قبل الولادة
يتم فحص الزوجين المقبلين على الحمل أو الراغبين فيه للبحث عن طفرات جين CFTR، لتحديد كافة احتمالات ولادة طفلهم بمرض التليف الكيسي. وعادًة ما يتم إجراء هذا الاختبار على واحد من الزوجين أو كليهما. وفي حالة الكشف عن زيادة خطر الإصابة بتليف كيسي، يتم إجراء هذا الاختبار على الجنين أيضًا. ويرجع ذلك إلى أن تشخيص ما قبل الولادة لا يُتيح نسب عالية أو بديلة من العلاج، والسبب الرئيسي لإجراء مثل هذا الفحص، من الناحية العملية، هو توفير إمكانية الإجهاض في حالة ما إذا كان الجنين حامًلا لهذا المرض. يُقدم اختبار الكشف عن التليف الكيسي على نطاق أوسع في بلاد كالولايات المتحدة.[39] وتُوصي الكلية الأمريكية لأطباء النساء والتوليد بإجراء الاختبار على الزوجين الذين لديهم تاريخ مرضي مع التليف الكيسي بين عائلاتهم وأقاربهم، لأن نسبة كبيرة من خطر الإصابة بالمرض يرجع سببه إلى الوراثة.[40]
ولأن تطور التليف الكيسي عند الجنين يتطلب من كلا الزوجين نقل نسخة من الجين الطافر CFTR، وبسبب التكلفة العالية لاختبار ما قبل الولادة، يتم إجرائه في البداية على أحد الأبوين دون الآخر. فإذا تبين أنه حامًلا لطفرة من جين CFTR، ففي هذة الحالة يتم فحص الآخر لتحديد خطر أن يُولد الطفل بهذا المرض. ويُمكن أن يحدث التليف الكيسي بسبب وجود أكثر من ألف طفرة مُختلفة. ففي عام 2006، لم يكن من المُمكن إجراء دراسات مختبرية سريرية لكل طفرة. ويتم إجراء هذا الاختبار لتحليل الدم بحثًا عن الطفرات الأكثر شيوعًا، مثل طفرة ΔF508، وقد كشفت معظم الأساليب المتوفرة ما لا يزيد عن 32 نوعًا مختلفًا. فإذا ما تم التعرف على التاريخ الذي حملت فيه عائلة ما نوع من الطفرات غير الشائع، فمن المُمكن حينئذ الكشف عن هذه الطفرة تحديدًا. ويُستنتج من ذلك أنه ليست كل الطفرات المعروفة تم الكشف عنها من خلال الاختبارات المُجْراه حديثًا، فالنتيجة السلبية للتحليل ليست ضامنًا بأن يُولد الطفل خاليًا من المرض.[41] وبالإضافة إلى ذلك، فإن الطفرات الأكثر شيوعًا التي تم اكتشافها هي بالضرورة الأكثر خطرًا، والاختبارات الوراثية الأقل خطرًا أقل نجاحًا، وبذلك فإن الطفرات الأكثر انتشارًا في هذه المجموعات هي أقل وجودًا في عامة السكان.
وفي كثير من الأحيان، يتم إجراء اختبارات إضافية أثناء الحمل أو قبله على الأزواج الأكثر عُرضة للخطر. ويُوفر التلقيح الصناعي في المختبر إلى جانب التشخيص الوراثي قبل عملية التلقيح إمكانية فحص الجنين قبل زراعته في الرحم. ويتم إجراء هذا الاختبار بعد مرور 3 أيام من عملية التخصيب، وهو يسعى إلى تحديد وجود جينات غير طبيعية من CFTR. فإذا تم تحديد اثنين من الجينات الطافرة من CTRF في الجنين، يتم استبعاده من عملية النقل، ويتم زراعة آخر يحمل على الأقل جين واحد طبيعي.
من المُمكن إجراء اختبارات خلال فترة الحمل على المشيمة، فحص الزغابات المشيمية، وعلى السائل الأمنيوسي الذي يحيط بالجنين، بزل السائل الأمنيوسي، وذلك باستخدام الموجات فوق الصوتية. ومع ذلك، فإن خزعة الزغابات المشيمية ترتبط بخطر موت الجنين بمعدل 1 في المائة، ومع بزل السلى بنسبة 1 في 200،[42] لذلك، لابُد من تحديد الفوائد بشكل صحيح لتحقيق التوازن بين المُخاطر قبل الشروع في الاختبار. وبدلاً من ذلك، يختار بعض الأزواج الخضوع لتقنيات التلقيح بالمساعدة بواسطة بويضات مأخوذة، اللجوء إلى التخصيب في المختبر، أو عن طريق عينات السائل المنوي المأخوذة، التلقيح الاصطناعي.
العلاج
أحد الجوانب الرئيسية في علاج التليف الكيسي هو السيطرة على التلف الرئوي الناجم عن سُمك المُخاط والالتهابات وذلك من أجل تحسين جودة حياة المريض. ولعلاج الالتهابات المُزمنة الحادة يتم أخذ مضادات حيوية عن طريق الحقن الوريدي والاستنشاق والفم. وأيضًا يتم استخدام الأجهزة الآلية والأدوية للسيطرة على الإفرازات، وبالتالي تُخفف من تراكم السوائل وتعمل على إبعادها عن مجرى التنفس. وهناك جوانب علاجية أخرى تتصل بعلاج مرض السكري بواسطة الأنسولين وأمراض البنكرياس بواسطة التعويض الإنزيمي. وبالإضافة إلى ذلك، يتم افتراض فعالية عدة إجراءات مختلفة، مثل: زراعة الأعضاء والعلاج الجيني، لحل بعض الآثار المرتبطة بهذا المرض. كما أن اتباع نظام غذائي صحي ومُمارسة الرياضة وعلاج الاضطرابات العدوانية بواسطة تناول المضادات الحيوية، يُزيد من متوسط عمر المرضي.
مضادات حيوية لعلاج المرض الرئوي[43]

يتم وصف المضادات الحيوية دائمًا عند الاشتباه في مرض ذات الرئة أو وجود خلل في وظائف الرئة. وعادًة ما يتم تحديد نوع المضادات الحيوية وفقًا لتاريخ إصابة المريض بالعدوى. فالعديد من أنواع البكتيريا المُسببة للتليف الكيسي لديها مُقاومة ضد الكثير من المضادات الحيوية، وبالتالي تحتاج لأسابيع من العلاج عن طريق الحقن الوريدي للفانكوميسين والتوبراميسين والميروبينيم والسيبروفلوكساسين والبيبيراسيلين.
فالعلاج طويل المفعول غالبًا ما يتطلب التواجد بالمشتفى بشكل دائم وذلك لإخد جُرعات العلاج عن طريق الوريد بشكل مُستمر مثل: القسطر المركزي الذي يتم إدخاله عن طريق الجلد. وأيضًا في كثير من الأحيان، يتم أخد جُرعات من المضادت الحيوية عن طريق الاستنشاق بشكل متوالٍ لعدة شهور مثل: التوبراميسين والكوليستين والجنتاميسين، وذلك من أجل تحسين وظائف الرئة ومنع انتشار البكتيريا.[44] ويتم استخدام بعض المضادات الحيوية الفموية أحيانًا مثل: السيبروفلوكساسين أو الأزيثروميسين، للمساعدة في منع العدوى والسيطرة عليها بمجرد انتشارها.[45]
وهناك العديد من المضادات الحيوية الشائعة مثل التوبراميسين والفانكومايسين، قد تُسبب فقدان السمع من خلال الإصابة بالتسميم الأذني والقصور الكلوي. ومن أجل منع هذة الآثار الجانبية، يُقاس عادًة نسبة تركيزات هذة الدوية في الدم لضبط الجرعة.
طرق أخرى لعلاج المرض الرئوي

هناك العديد من التقنيات التي يتم تنفيذها بهدف إذابة البلغم وتسهيل طرده. ففي المستشفيات، يُستخدم العلاج الفيزيائي، حيث يمارس أخصائي العلاج سلسلة من المناورات بواسطة الضغط والقرع على الصدر من الخارج عدة مرات في اليوم. فالأجهزة الميكانيكية تعمل تحت نفس المبدأ الذي تقوم عليه الآليات الأساسية ألا وهو التجفيف والنزح الوضعي، ومن بينهم يُوجد أجهزة تهوية عالية التذبذب وأجهزة تهوية طرقية تعمل داخل الرئة، ومنهم أيضًا المحمول المهيأ للاستخدام المنزلي.[46] والتمارين الرياضية مُفيدة للغاية بالنسبة لمرضى التليف الكيسي، لأنها ليست فقط تعمل على تخفيف تراكم البلغم، ولكنها أيضًا تُحسن من صحة القلب والأوعية الدموية والصحة العامة.

من بين العقاقير التي يتم أخذها عن طريق الاستنشاق، حيث تعمل على التخفيف من الإفرازات البلغمية وتُسهل طردها، هناك دورناز ألفا والمحلول الملحي مفرط التوتر.[47] فالدورناز ألفا هو عامل إنزيمي يُسمى ديوكسي، أدنازا أو دنازا، البشري المأشوب، وهو يعمل على تفكيك الحمض الريبوزي النووي منزوع الأوكسجين في البلغم، وبذلك يقلل من لزوجته.[48] ويعمل أسيتيل سيستئين، وهو مُشتق من الحمض الأميني سيستئين، على إذابة البلغم، وقد أظهرت الأبحاث والتجارب المُتاحة قلة النتائج المُترتبة عليهم. وأخيرًا، فإن موسعات القصبات الهوائية مثل السالبوتامول والسالميتيرول، وكلاهما محاكي الودي أو ناهضاتβ2- للأدرينالين، أو الإبراتروبيوم بروميد، ناهضة لمستقبل أستيل كولين، مُشتق رباعي من الأتروبين، يتم استخدامهم لتوسيع حجم المسالك التنفسية الضيقة عن طريق إرخاء العضلات الملساء للقصبة الهوائية.
فكلما ازدادت الحالة الرئوية سوءًا، كلما تطلب ذلك دعم التهوية الألية. ويتعين على بعض المرضى ارتداء أقنعة التنفس المُخصصة لهم أثناء النوم لأنها تعمل على تدفق الهواء إلى الرئتين. وتُساعد التهوية غير الغازية، التي تتم من خلال إعطاء الأكسجين تحت ضغط إيجابي متقطع عن طريق قناع الأنف، VPAP وهو اختصار إنجليزي لمُتغير ضغط الهواء الإيجابي، على منع هبوط مستويات الأكسجين في الدم أثناء النوم. ويُمكن أيضَا أن تُستخدم أثناء دورة العلاج الطبيعي للصدر لتعزيز طرد البلغم.[49] وعلى الرغم من ذلك، قد يكون من الضروري في حالات الطوارئ استخدام التهوية الغازية مع التنبيب الرغامي، وهو عبارة عن وضع أنبوب أو قسطر في القصبة الهوائية.
علاج جوانب أخرى من التليف الكيسي

عادًة ما يتطلب التدخل الجراحي في حالة إصابة الأطفال الرضع بالعلوص العِقْي، ولا يحدث هذا غالبًا عند البالغين الذين يُعانون من متلازمة الانسداد المعوي. ويقوم علاج قصور البنكرياس على أساس إيجاد بديل عن الإنزيمات الهضمية التي تسمح للأمعاء بامتصاص العناصر الغذائية والفيتامينات التي يتم فقدانها في البراز بشكل مناسب. ومع ذلك، ينبغي على الأشخاص المُصابين بالتليف الكيسي أخذ جرعات زائدة من فيتامين ألف ودي وإي وك، بداية من المكملات الغذائية واتباع نظام غذائي عالي السعرات الحرارية. وعادًة ما يُصاحب التليف الكيسي مرض السكري الذي يتم معالجته عن طريق حقن الإنسولين.[50] ويُمكن الوقاية من سرعة تطور مرض هشاشة العظام عن طريق تناول مُكملات غذائية من فيتامين دي والكالسيوم، ولكن عادًة ما يتم معالجتها من خلال عقار بيسفوسفونات.[51] وفيما يتعلق بتأخر النمو، فمن المُمكن الحد منه عن طريق إدخال أنبوب تغذية إلى داخل المعدة، عملية فغر المعدة، لتغذية المريض، وبالتالي زيادة امتصاص السعرات الحرارية من خلال التغذية الإضافية، وأيضًا يُمكن التعاطي لهذا الغرض حقن هرمون النمو.[52]
يتم عادًة مُعالجة الجيوب جانب الأنفية باستخدام جرعات المضادات الحيوية طويلة المفعول. فتزايد نمو اللحمِيات داخل الممرات الأنفية، يُشبه باقي التغيرات الهيكلية المرضية، ويُمكنه أن يُقيد تدفق الهواء إلى داخل الرئة. ولهذا السبب، يتطلب التدخل الجراحي في محاولة لتخفيف الانسداد الذي تسببه والحد من حدوث التهابات جديدة. وأيضًا، يتم أخذ دواء كورتيكوستيرويد داخل الأنف ودواء فلوتيكازون للحد من الالتهاب.[53] ومن ناحية أخرى، يُمكن مُحاولة التصدي لمُشكلة العقم عند النساء من خلال اللجوء لتقنيات التلقيح الصناعي. ويُمكن مُعالجة هذه المشكلة عند الرجال أيضًا عن طريق حقن الحيوانات المنوية داخل الهيلولي.[54]
زراعة الرئة وتنظيم بروتين CFTR والعلاج الجيني[55]
بشكل عام، يُعتبر من الأفضل زراعة الرئة لدى الأشخاص الذين يُعانون من تدهور تدريجي في وظائف الرئة وشدة الإجهاد بعد ممارسة الرياضة، حيث تعب وإرهاق عضلي شديد غير متناسب مع كم التمارين. وبالرغم من أن المُجدي في كثير من الأمراض زرع رئة واحدة فقط، إلا أنه يجب استبدال كلا الرئتين عند الأشخاص المُصابين بالتليف الكيسي، لأن البكتيريا العالقة بباقي العضو يُمكنها أن تصيب الرئة المزروعة. ومن الممكن أيضًا إجراء عملية زرع بنكرياس وكبد بشكل متتالي للحد من مرض الكبد والسكرى.[56] ويتم البدء في تقييم فكرة زراعة الرئة في حالة وجود خلل في الوظيفة الرئوية يُهدد حياة المريض أو في حالة عدم قدرة المريض على التنفس إلا من خلال الأجهزة الآلية.[57]

يُعتبر العلاج الجيني وسيلة فعالة في التصدي لمرض التليف الكيسي. بحيث يتم من خلاله إدخال نسخة سليمة من جين CFTR إلى الخلايا المُتضررة. ونظرًا إلى عدم قدرة الفيروسات القهقرية على تمكين الخلايا من الانقسام الخلوي، فوجب إجراء تحاليل سريرية لإدخال الجينات في الفيروسات الغذانية. وفي الوقت الراهن، تُستخدم هذه الفيروسات في الفحوصات التي يتم فيها حقن جين CFTR السليم إلى الخلايا الظهارية المُبطنة للرئتين عن طريق استخدام أسلوب الهباء الجوي، إحدى العلاجات الجينية التي يتم استخدامها. ومن المُتوقع أن ينتج عن دخول الفيروسات الغدانية إلى الجين السليم وظيفة تتعلق بقنوات الكلور في هذه الخلايا.
أشارت بعض الدراسات إلى أنه للتمكن من منع المظاهر الرئوية لمرض التليف الكيسي، فإن ذلك يتطلب إجراء تعبير جيني ما بين 5 و10٪ من القيم السليمة من بروتين CFTR؛[58] حيث تكمن عوائق الفيروسات الغدانية في عدم دخولها في الحمض الريبوزي النووي المنزوع الأوكسجين المضيفة. وبالتالي يتم فقدانها مُسببة ذلك تعبير جيني مؤقت مع الحاجة إلى إعادة إدخال الناقل. وقد تم اقتراح اتباع أساليب مُختلفة وتم البدء في العديد من الدراسات السريرية. ولكن بحلول عام 2006، كانت العديد من العقبات ما زالت قائمة والتي سيكون من الضروري التغلب عليها لإثبات نجاح العلاج الجيني.[59]
يأتي النهج الأخر الذي يُتبع في علاج التليف الكيسي عن طريق استخدام معززات ومُحسنات أو منظمات لبروتين CFTR لإصلاح الخلل الكامن في تكوين هذا البروتين. ووُجدت العديد من العقارات المعززة في مراحل مختلفة من الأبحاث، ومن بينهم: عقار إيفا كافتور ولاما كافتور وتيزا كافتور. وفي البداية، تم تخصيص عقار إيفا كافتور المُحسن، كان يُطلق عليه اسم VX770 تحت الاسم التجاري Kalydeco، للمرضى الذين تجاوزت أعمارهم 6 أعوام. وبداية من عام 2015، تم تمكين المرضى الذين تتراوح أعمارهم من 2 إلى 5 أعوام من أخذ العقار وذلك وفقًا لما صرحت به إدارة الغذاء والدواء.[60] وقد صدر هذا التصريح من أجل علاج طفرة G551D، وذلك لإصابة أقل من 3% من إجمالى مرضي التليف الكيسي بها.[61] وفي عام 2014، صرحت إدارة الغذاء والدواء بإمكانية استخدام هذا العقار أيضًا في علاج طفرات G178R وS549N وS549R وG551S وG1244E وS1251N وS1255P وG1349D.[62] وتم تحسين FEV1 بشكل عام بنسبة 10.4%.[63] وقد تجاوزت تكلفة هذا العلاج 000.300 دولار أمريكي للمريض الواحد في عام 2014. وتمت الموافقة من قِبل إدارة الغذاء والدواء والاتحاد الأوروبي على الدمج بين عقار إيفا كافتور ولاما كافتور، كان يُسمي سابقًا VX-809، في مختبر فيرتكس للأدوية، والسماح للمصابين بطفرة AF508، الشكل الأكثر انتشارًا، بأخده في حالة تماثل الألائل، وهي نسختين مُتماثلتين من المورثة.[64] وفي الوقت الحالي، يتم إجراء محادثات مع مختلف دول الاتحاد الأوروبي للتفاوض بشأن سداد قيمة تكاليف الأدوية.[65] وهناك أيضًا في إطار البحث، الجمع بين عقار إيفا كافتور مع المعدل من دواء تيزا كافتور، كان يُطلق عليه سابقًا VX-661، بالنسبة للأشخاص الذين يُعانون من طفرة F508 في حالة تغاير الألائل، وهي نسخة مختلفة من كل أليل.[66]
وفيما يتعلق بطفرات النوع الأول 1، وتُسمى أيضًا بالطفرات اللاشعورية، فهي تتواجد في الكودون الختامي الذي من شأنه أن يوقف عملية تشكيل البروتين في مرحلة مُبكرة، وبالتالي لا يقوم بوظيفته، وقد تم اختبار الأدوية التي أظهرت بعض فاعليتها في قدرة الريبوسومات على تجاهل الكودونات مُبكرًا وتكوين بروتين CFTR. ووفقًا لما صرحت به إدارة الغذاء والدواء، فإن عقار أتالورين، كان يُسمى PTC124، لعلاج مرض ضمور العضلات في مختبرات بي تي سي ثيرابويتكس وصل إلى نتائج غامضة في المرحلة الثالثة من البحث بالنسبة لغالبية المرضى الذين يعانون من هذه الطفرات، على الرغم من أن نفس النتائج مُبشرة بالنسبة للمرضى الذين لا يُعانون من مرض مزمن بسبب المضادات الحيوية.[67]
وتسعى أنواع أخرى من الأدوية إلى فتح قنوات بديلة للكلور في الخلية، ولكن ما زال البحث والتجارب في مراحل مبكرة.
علم الأوبئة

التليف الكيسي هو الأكثر انتشارًا من بين الأمراض الصبغية الجسمية المُتنحية بين الأفراد الذين تعود جذورهم إلى أصول أوروربية، ويُؤدي إلى الوفاة في بعض الحالات. ففي الولايات المتحدة، بلغ عدد المصابين بالتليف الكيسي ما يقرب من 30000 شخص، وفي الغالب يتم تشخيصهم في عُمر الستة أشهر. وفي كندا، بلغت أعدادهم 3000 فرد. وتُشير الإحصائيات إلى أن واحد من كل 25 فرد من أصل أوروبي وواحد من كل 29 شخص من أصل أشكنازي حاملين لطفرة التليف الكيسي. وعلى النقيض، يقل انتشار المرض بين هذه المجموعات، فتقريبًا شخص من كل 46 من أصول أسبانيا، وفرد من كل 65 أفريقي وشخص من كل 90 آسيوي يكون حامًلا على الأقل لجين واحد من CFTR غير سليم.[68][69][70] وتُعتبر كًلا من الأرجنتين والأوروغواي استثناءً من بين دول أمريكا اللاتينية، وذلك بسبب وقوع حالات وبائية أكثر بكثير من متوسط حالات المنطقة، فهي قريبة جدًا من الإحصائيات التي تم رصدها في الولايات المتحدة وكندا، وبذلك انتشرت فرق الحملات الصحية بين التجمعات السكنية.[71]
يتم تشخيص التليف الكيسي في الرجال والنساء. ولأسباب غير معروفة كليًا، فإن متوسط العمر المتوقع عند الولادة يزيد بين المواليد الذكورالمُصابين عن الإناث.[72] ويتفاوت هذا المؤشر بشكل كبير بحسب نطاق ونوعية الرعاية التي تقدمها نُظم الصحة العامة. ففي عام 1959، بلغ متوسط عمر الأطفال المُصابين بالتليف الكيسي 6 أشهر. وفي عام 2006 في الولايات المتحدة الأمريكية، ارتفع متوسط العمر ليصل إلى 36.8 عام، وذلك وفقًا للبيانات التي أحصتها مؤسسة التليف الكيسي.[73] وقد ارتفع متوسط العمر المتوقع بشكل مماثل في أجزاء كبيرة من الغرب، باستثناء الدول الأقل تطورًا، فقد تم إحصاء أرقام أقل بكثير من ذلك، حيث أن الغالبية العُظمى المُصابة لا تتخطى أعمارهم 10 سنوات.
قامت مؤسسة التليف الكيسي بإحصاء معلومات عن نمط حياة البالغين المصابين بالتليف الكيسي في الولايات المتحدة الأمريكية. وفي عام 2004، أفادت المؤسسة بإن 91٪ من هذة الفئة السكانية أتموا بالفعل مرحلة التعليم الثانوي، و54% تم قبولهم في الجامعات. وكشفت البيانات المُتعلقة بفئة العمال المُصابين بأن 12.6% من هؤلاء البالغين ليست لديهم القدرة على العمل، حيث تركوا نطاق القوى العاملة)، و9.9% يُعانون من البطالة. ومن ناحية أخرى، أشارت الإحصائيات الزوجية بأن 59% من المصابين كان أعزب و36% كان متزوجًا أو يعيش مع رفيقة. وفي عام 2004 في الولايات المتحدة الأمريكية، بلغ عدد النساء الحوامل المُصابات بالتليف الكيسي 191.[74]
نظريات حول انتشار مرض التليف الكيسي
تُشير النظريات إلى أن الطفرة AF508 مُتواجدة منذ 52000 سنة.[75] وقد وُضعت العديد من الفرضيات التي تحاول أن تشرح سبب انتشار هذه الطفرة المميتة بين جموع السكان. فبعض الأمراض الصبغية الجسمية المُتنحية المُنتشرة مثل فقر الدم المنجلي أثبتت حماية حامل المرض من الأمراض الأخرى، وهو مفهوم يُعرف بالميزة المُتغايرة. ومع اكتشاف أن ذيفان الكوليرا يحتاج إلى بروتينات CFTR سليمة ليعمل بشكل صحيح، تم افترض أن ناقلات الجينات الطافرة من CFTR تم الحصول عليها من أجل مُقاومة مرض الكوليرا وغيرها من مسببات الإسهال.[76] ومع ذلك، لم تُؤكد الدراسات الحديثة هذه الفرضية.[77][78]
فوجود بروتينات طبيعية من CFTR شرط ضروري لدخول بكتيريا السالمونيلا التيفية، نمط مصلي من سلمونيلة كوليرا الخنازير، وهي ضمن شُعبة المتقلبات، وهي بكتيريا سلبية الغرام من جنس السلمونيلا) في الخلايا،[79] ويُشير ذلك إلى أن ناقلات جينات CFTR الطافرة يُمكن أن تكون مُقاومة لحمى التيفوئيد. ومع ذلك، لم تُؤكد أية دراسة مُجراة هذه الافتراضية. وفي كلتا الحالتين، فقد انخفض معدل وقوع إصابات بمرض التليف الكيسي خارج أوروبا، في مناطق توطن أمراض الكوليرا وحمى التيفوئيد، ولكن ذلك يفتقر أيضًا إلى تفسير عاجل.
تاريخ المرض

لم يتم التعرف على الطيف السريري الكامل لمرض التليف الكيسي حتى عقد 1930، ولكن بعد ذلك تم تحديد بعض الجوانب منه. وقد وصف الطبيب كارل بون روكيتانسكي حالة وفاة جنين بسبب إصابته بالتهاب الصفاق بالعقي، بأن مضاعفات العلوص العقي مُرتبطة بالتليف الكيسي. وقد تم وصف العلوص العقي لأول مرة عام 1905 من قِبل الطبيب كارل لاندشتاينر.[80]
في عام 1938، نشرت الطبيبة دوروثي هانسين أندرسون مقالًا في الجريدة الأمريكية لأمراض الأطفال بعنوان التليف الكيسي للبنكرياس وعلاقته بأمراض الجهاز الهضمي، دراسة سريرية ومرضية. وبهذا المقال، كانت أول باحثة تُعْرف وتُصنف هذا الكيان المرضي، المُسمى في هذا الوقت، بالتليف الكيسي للبنكرياس، وتربطه بالاضطرابات الرئوية والمعوية.[3] وافترضت أيضًا كونه مرض مُتنح، وتم استخدام التعويض عن إنزيمات البنكرياس كعلاج للأطفال المُصابين. وفي عام 1952، اكتشف الطبيب بول دي سانت خللًا في شوادر العرق، وبناءًا على هذه الأدلة، تم تطوير اختبار العرق خلال العقد الذي يليه.[81]
في عام 1985، رسم باحثون من لندن ومن تورونتو وسولت ليك خريطة جين CFTR على الكروموسوم 7q. وبعد أربع سنوات، وفي عام 1989، اكتشف علماء الجينات فرانسيس كولينز ولاب شي تسوي وجون ريوردان أول طفرة للتليف الكيسي AF508، التي تقع على نفس الكروموسوم. وقد حددت الأبحاث التي جاءت بعد هذا الاكتشاف أكثر من ألف طفرة مُختلفة تؤدي إلى هذا المرض. وكان العالم لاب شي تسوي يقود فريق من العلماء في مُستشفى للمرضى الأطفال، إحدى المُستشفيات التعليمية باتفاق مع جامعة تورنتو، والذي اكتشف الجين المسئول عن التليف الكيسي. وهو يُعتبر أول اضطراب جيني تم توضيحه بدقة من خلال عملية الوراثة العكسية. ويرجع ذلك إلى أن طفرات جين CFTR عادًة ما تكون صغيرة، ولم تكن التقنيات القديمة لعلم الوراثة قادرة على تحديد دقيق للجين الطافر. وباستخدام العوامل البروتينية، تمكنت دراسات الربط الجيني من رسم خريطة للطفرة على الكروموسوم 7. وقد ساعدت تقنيات الكروموسومات المشي والقفز على تحديد وتسلسل الجين. وكان هذا الجين من أوائل الجينات التي تم تحديد موقعهم وتسلسلهم بواسطة الخريطة الجينية، وكان فرانسيس كولينز من المشاركين في مشروع الجينوم البشري.
فالتعرف على طفرة معينة مسئولة عن التليف الكيسي عند المريض قد يكون مُفيدًا في التنبؤ بتطور المرض. فعلى سبيل المثال، يُعاني المرضى متماثلو الألائل في الطفرة AF508، في معظم الحالات تقريبًا، من قصور في البنكرياس والتهابات حادة في الجهاز التنفسي. وبالرغم من ذلك، يوجد بعض الاحتمالات التي تشير إلى هناك عوامل إضافية، رُبما تكون جينات في موقع كروموسومي آخر، تُشارك في التعبير عن المرض. ومن ناحية أخرى، أتاح استنساخ جين التليف الكيسي، إمكانية العلاج الجيني، كما هو موضح في القسم المختص.
انظر أيضًا
مراجع
- التليُّف الكيسي نسخة محفوظة 18 مايو 2017 على موقع واي باك مشين.
- التليّف الكيسي عند الأطفال: الأعراض والعلاج نسخة محفوظة 01 أغسطس 2017 على موقع واي باك مشين.
- Andersen DH. "Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study." Am J Dis Child 1938; 56:344-399
- Rosa María Girón Moreno y Antonio Salcedo Posadas Fibrosis quística. Monografías Neumomadrid. Volumen VIII / 2005. [1] Edita: ERGON. C/ Arboleda, 1. 28220 Majadahonda (Madrid). (15 de diciembre de 2005).
- Yankaskas JR, Marshall BC, Sufian B, Simon RH, Rodman D. (2004). «Cystic Fibrosis adult care consensus conference report»[وصلة مكسورة]. Chest 125: 1-39. doi:10.1378/chest.125.1_suppl.1S. PMID 14734689. نسخة محفوظة 25 أبريل 2015 على موقع واي باك مشين. [وصلة مكسورة]
- Isabel Largo García (2009). «Fibrosis quística». Revista Pediatría Electrónica 6. ISSN 0718-0918. نسخة محفوظة 13 مارس 2016 على موقع واي باك مشين.
- Jorde, Lynn; Carey, John; White, Raymond. Genética médica. Madrid: Mosby, 1996. ISBN 84-8174-161-2
- New Statistics Show CF Patients Living Longer Cystic Fibrosis Foundation (26 de abril، 2006). Consultado el 26-07-2006. نسخة محفوظة 07 أكتوبر 2015 على موقع واي باك مشين. [وصلة مكسورة]
- Canadian Cystic Fibrosis Patient Data Registry Report. Cystic Fibrosis Canada, 2010. Consultado el 21 de diciembre de 2012.
- أعراض الإصابة بالتليف الكيسي نسخة محفوظة 16 أغسطس 2017 على موقع واي باك مشين.
- التليف الكيسي نسخة محفوظة 22 فبراير 2014 على موقع واي باك مشين.
- Rowe SM, Miller S, Sorscher EJ. «Cystic Fibrosis.» N Engl J Med. 2005 may 12;352(19):1992-2001. PMID 158887000.
- Maldonado M, Martínez A, Alobid I, Mullol J. «The antrochoanal polyp.» Rhinology. 2004 dic;42(4):178-82. Rev. PMID 15626248.
- Ramsey B, Richardson MA. «Impact of sinusitis in cystic fibrosis.» Allergy Clin Immunol. 1992 sep;90(3 Pt 2):547-52. PMID 1527348.
- Eggermont E, De Boeck K. «Small-intestinal abnormalities in cystic fibrosis patients.» Eur J Pediatr. 1991 oct;150(12):824-8. Rev. PMID 1743211
- Sun L, Rommens JM, Corvol H, Li W, Li X, Chiang TA, Lin F, Dorfman R, Busson PF, Parekh RV, Zelenika D, Blackman SM, Corey M, Doshi VK, Henderson L, Naughton KM, O'Neal WK, Pace RG, Stonebraker JR, Wood SD, Wright FA, Zielenski J, Clement A, Drumm ML, Boëlle PY, Cutting GR, Knowles MR, Durie PR, Strug LJ. «Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis.» Nat Genet. 2012 Apr 1. PMID 22466613.
- Kulczycki LL, Shwachman H. "Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse." N Engl J Med. 1958 ag 28;259(9):409-12. PMID 13578072
- Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998 sep 3;339(10):653-8. PMID 9725922
- Malfroot A, Dab I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child. 1991 nov;66(11):1339-45. PMID 175564
- Khoshoo V, Udall JN Jr. Meconium ileus equivalent in children and adults. Am J Gastroenterol. 1994 feb;89(2):153-7. PMID 8304294
- Williams SG, Westaby D, Tanner MS, Mowat AP. Liver and biliary problems in cystic fibrosis. Br Med Bull. 1992 oct;48(4):877-92. PMID 1458306
- Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER. "Insulin sensitivity in cystic fibrosis." Diabetes. Agosto 1994;43(8):1020-6. PMID 8039595
- Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, Economou G, Horrocks AW, Freemont AJ, Mawer EB, Adams JE. "Low bone mineral density in adults with cystic fibrosis." Thorax. 1999 nov;54(11):961-7. PMID 10525552
- Gibson LE, Cooke RE. "A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine by iontophoresis." Pediatrics mar;23(3):545-9. PMID 13633369
- Stern, RC. "The diagnosis of cystic fibrosis." N Engl J Med 1997; 336:487. PMID 9017943
- Saiman L. Microbiology of early CF lung disease. Paediatr Respir Rev. 2004;5 Supl A:S367-9. PMID 14980298
- Tummler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H. Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients. J Clin Microbiol. 1991 jun;29(6):1265-7. PMID 1907611
- Centers for Disease Control and Prevention (CDC). Pseudomonas cepacia at summer camps for persons with cystic fibrosis. MMWR Morb Mortal Wkly Rep. 1993 jun 18;42(23):456-9. PMID 7684813
- Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR.Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group. J Pediatr. 1994 may;124(5 Pt 1):694-702. PMID 7513755
- Pankhurst CL, Philpott-Howard J. The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients. J Hosp Infect. 1996 Apr;32(4):249-55. PMID 8744509
- Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK. Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak. Thorax. 2003 jun;58(6):525-7. PMID 12775867
- Hoiby N. Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa. Neth J Med. 1995 jun;46(6):280-7. PMID 7643943
- Blaschke et al. Mycologucal surveillance of chlidren wiyh cystic fibrosis. Mycoses. 1991;34 Suppl 1:43-7
- Maiz L et al. Serologic IgE inmune responses against Aspergillus fumigatus and Candida albicans in patient with cystic fibrosis. Chest 2002;121:782-8
- «Cystic Fibrosis Mutation Database: Statistics». Genet.sickkids.on.ca. 25 de abril de 2011. Consultado el 17 de diciembre de 2011. نسخة محفوظة 01 أغسطس 2017 على موقع واي باك مشين.
- Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998 jul 31;273(31):19797-801. PMID 9677412 نسخة محفوظة 07 نوفمبر 2017 على موقع واي باك مشين.
- التليف الكيسي نسخة محفوظة 31 أكتوبر 2015 على موقع واي باك مشين.
- "Cystic Fibrosis Mutation Database: Statistics"، مؤرشف من الأصل في 30 يوليو 2018.
- .American College of Obstetricians and Gynecologists and American College of Medical Genetics. Preconception and prenatal carrier screening for cystic fibrosis. Clinical and laboratory guidelines. American College of Obstetricians and Gynecologists, Washington, DC, octubre 2001
- Elias, S, Annas, GJ, Simpson, JL. Carrier screening for cystic fibrosis: Implications for obstetric and gynecologic practice. Am J Obstet Gynecol 1991; 164:1077. PMID 2014829
- Tabor A, Philip J, Madsen M, Bang J, Obel EB, Norgaard-Pedersen B. Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet. 1986 jun 7;1(8493):1287-93. PMID 2423826
- Pai VB, Nahata MC. Efficacy and safety of aerosolized tobramycin in cystic fibrosis. Pediatr Pulmonol. 2001 oct;32(4):314-27. Rev. PMID 11568993
- تليف كيسي نسخة محفوظة 24 أكتوبر 2016 على موقع واي باك مشين.
- Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J Cyst Fibros. 2004 mar;3(1):23-8. PMID 15463883
- Hansen CR, Pressler T, Koch C, Hoiby N.Long-term azithromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study. J Cyst Fibros. 2005 mar;4(1):35-40. PMID 15752679
- Kuver R, Lee SP. Hypertonic saline for cystic fibrosis. N Engl J Med. 2006 abr 27;354(17):1848-51; respuesta del autor 1848-51. PMID 16642591
- Lieberman J. "Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis." JAMA. 1968 jul 29;205(5):312-3. PMID 5694947
- Moran F, Bradley J. Non-invasive ventilation for cystic fibrosis. Cochrane Database Syst Rev. 2003;(2):CD002769. Rev. PMID 12804435
- Onady GM, Stolfi A. Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst Rev. 2005 jul 20;(3):CD004730. Rev. PMID 16034943
- Conway SP, Oldroyd B, Morton A, Truscott JG, Peckham DG. Effect of oral bisphosphonates on bone mineral density and body composition in adult patients with cystic fibrosis: a pilot study. Thorax. 2004 ag;59(8):699-703. PMID 15282392
- Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, Prestidge C, Seilheimer DK, Shepherd R. Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition.J Pediatr. 2005 mar;146(3):324-8. PMID 15756212
- Marks SC, Kissner DG. Management of sinusitis in adult cystic fibrosis. Am J Rhinol. 1997 en-feb;11(1):11-4. PMID 9065342
- Phillipson GT, Petrucco OM, Matthews CD. Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection. Hum Reprod. 2000 feb;15(2):431-5. PMID 10655317
- Simultaneous liver and pancreas transplantation in patients with cystic fibrosis. Transplant Proc. 2005 oct;37(8):3567-9. PMID 16298663
- أعراض وعلاج التليف الكيسي نسخة محفوظة 23 يوليو 2016 على موقع واي باك مشين.
- Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, Yee JY, Kotloff RM, Lipson DA, Bunin GR. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2006 mar 15;173(6):659-66. Epub 2005 dic 30. PMID 16387803
- Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002 nov;27(5):619-27. PMID 12397022
- Tate S, Elborn S. Progress towards gene therapy for cystic fibrosis.Expert Opin Drug Deliv. 2005 mar;2(2):269-80. Rev. PMID 16296753
- "Vertex Receives U.S. Food and Drug Administration Approval of KALYDECO® (ivacaftor) for Children with Cystic Fibrosis Ages 2 to 5 who have Specific Mutations in the CFTR Gene" (باللغة الإنجليزية)، مؤرشف من الأصل في 09 مارس 2018، اطلع عليه بتاريخ 16 يوليو 2017.
- I nforme de Posicionamiento Terapéutico de Ivacaftor (Kalydeco®). Ministerio de Sanidad Servicios Sociales e Igualdad (España), publicado el 20 de junio de 2014. Consultado el 7 de julio de 2014. [وصلة مكسورة] نسخة محفوظة 5 يوليو 2016 على موقع واي باك مشين.
- «U.S. Food and Drug Administration Approves KALYDECO™ (ivacaftor) for Use in Eight Additional Mutations that Cause Cystic Fibrosis». investors.vrtx.com. Consultado el 10 de febrero de 2017. [وصلة مكسورة] نسخة محفوظة 22 مارس 2020 على موقع واي باك مشين.
- http://www.ema.europa.eu/docs/es_ES/document_library/EPAR_-_Summary_for_the_public/human/002494/WC500130744.pdf.
- «CF Therapy Orkambi Approved in Europe». www.cff.org. Consultado el 10 de febrero de 2017. نسخة محفوظة 08 سبتمبر 2018 على موقع واي باك مشين.
- «L a combinación lumacaftor e ivacaftor, de Vertex, mejora la función pulmonar en fibrosis quística». Redacción Médica. Consultado el 10 de febrero de 2017. نسخة محفوظة 11 فبراير 2017 على موقع واي باك مشين.
- (PDF) https://web.archive.org/web/20180908164543/http://files.shareholder.com/downloads/VRTX/3782532527x0x923399/A5C20E73-F92D-4BC7-8DD1-BF5152C86E7D/VRTX_Presentation_1-9-17.pdf، مؤرشف من الأصل (PDF) في 8 سبتمبر 2018، اطلع عليه بتاريخ أغسطس 2020.
{{استشهاد ويب}}: تحقق من التاريخ في:|تاريخ الوصول=(مساعدة)، الوسيط|title=غير موجود أو فارغ (مساعدة) - "Cystic Fibrosis Foundation - Drug Development Pipeline"، مؤرشف من الأصل في 18 يوليو 2015.
- Rosenstein BJ and Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998 abr;132(4):589-95. Rev. PMID 9580754
- Hamosh A, Fitz-Simmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998 feb;132(2):255-9. PMID 9506637
- Kerem B, Chiba-Falek O, Kerem E. Cystic fibrosis in Jews: frequency and mutation distribution. Genet Test. 1997;1(1):35-9. Rev. PMID 10464623
- (PDF) https://web.archive.org/web/20150924103512/http://www.smu.org.uy/publicaciones/sermedico/2011/sm3/dossier.pdf، مؤرشف من الأصل (PDF) في 24 سبتمبر 2015.
{{استشهاد ويب}}: الوسيط|title=غير موجود أو فارغ (مساعدة) - Rosenfeld, M, Davis, R, FitzSimmons, S, et al Gender gap in cystic fibrosis mortality. Am J Epidemiol 1997 145,794-803
- New Statistics Show CF Patients Living Longer Cystic Fibrosis Foundation (26 de abril، 2006). Consultado el 27-07-2006. نسخة محفوظة 07 أكتوبر 2015 على موقع واي باك مشين. [وصلة مكسورة]
- Cystic Fibrosis Foundation data (PDF) Consultado el 27/07/2006. [وصلة مكسورة] نسخة محفوظة 06 مارس 2016 على موقع واي باك مشين.
- Wiuf C. Do delta F508 heterozygotes have a selective advantage? Genet Res. 2001 ag;78(1):41-7. PMID 11556136
- Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994 oct 7;266(5182):107-9. PMID 7524148
- Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ. The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol. 1995 enero 15;482 (Pt 2):449-54. PMID 7714835
- Hogenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet. 2000 dic;67(6):1422-7. Epub 2000 oct 30. PMID 11055897
- Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998 may 7;393(6680):79-82. PMID 9590693
- Busch R. "On the history of cystic fibrosis." Acta Univ Carol [Med] (Praga). 1990;36(1-4):13-5. PMID 2130674
- Di Sant' Agnese PA, Darling RC, Perera GA, et al. «Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease.» Pediatrics 1953; 12:549-563.
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 sep 8;245(4922):1059-65. PMID 2772657
مصادر
- Dapena Fernández, Francisco Javier. Fibrosis quística. Salobreña: Alhulia, 1998, 1.ª ed. ISBN 84-95136-13-9
- Salcedo Posadas, Antonio; García Novo, María Dolores. Fibrosis quística. Madrid: Díaz de Santos, 1998, 1.ª ed. ISBN 84-7978-368-0
- Segal, Edgardo. Fibrosis quística. Buenos Aires: Journal, 2004, 1.ª ed. ISBN 987-97739-7-7
- (Segal E, et al. "Consenso de Fibrosis Quística." Arch.argent.pediatr. 1999;97(3):188. Disponible en línea (PDF
- Breve revisión de la genética de la Fibrosis Quística
وصلات خارجية
- ما هو التليف الكيسي وما خصائصه الجمعية الأندلسية للتليف الكيسي.
- بروتوكول تشخيصي وعلاجي عن التليف الكيسي بواسطة هيكتور إسكوبار وأمايا سوجو، الجمعية الإسبانية لطب الأطفال.
- التهابات الجهاز التنفسي الناجمة عن التليف الكيسي بواسطة خوسيه غونزاليس فالديز وغلاديس أبرو سواريز. القانون الطبي 2000;9(1-2):39-43.
- الإجماع الوطنى للتليف الكيسي بواسطة اجناسيو سانشيز وأنخليكا بيريز ولينا بوزا وآخرون لعام 2001; 72(4):356-80. ISSN 0370-4106.
بالإنجليزية
- اضطرابات ذات صلة بجين CFTR (جينريفيوس، مكتبة الطب الوطنية الأمريكية، NIH)
- التليف الكيسي في مدلاين MEDLINE
- التليف الكيسي في OMIM
- البوابة الرئيسية لعلم الوراثة مزودة بمجموعة معلومات متنوعة عن هذا المرض، وقد صدر هذا المحتوى من قبل معاهد الصحة الوطنية الأمريكية (تم النشر تحت بند الملكية العامة)
- مؤسسة جنيف للتعليم الطبي والبحوث. صور ذات صلة بالتليف الكيسي. جامعة جنيف، سويسرا.
- التليف الكيسي
- VX-770 إيفا كافتور، علاج فعال في المرحلة الثالثة (2011).
- تاريخ زراعة الرئة
 بوابة تمريض
بوابة تمريض بوابة طب
بوابة طب بوابة علم الأحياء التطوري
بوابة علم الأحياء التطوري بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي