Ciliopathy
| Ciliopathy | |
|---|---|
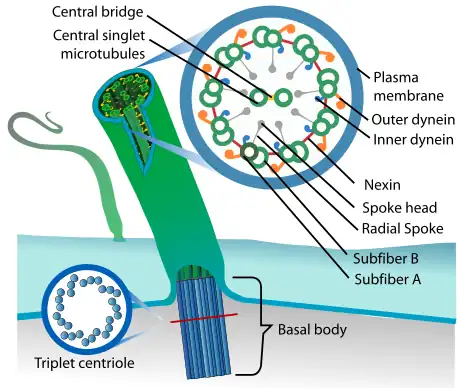 | |
| Eukaryotic cilium | |
| Specialty | Medical genetics |
A ciliopathy is any genetic disorder that affects the cellular cilia or the cilia anchoring structures, the basal bodies,[1] or ciliary function.[2] Primary cilia are important in guiding the process of development, so abnormal ciliary function while an embryo is developing can lead to a set of malformations that can occur regardless of the particular genetic problem.[3] The similarity of the clinical features of these developmental disorders means that they form a recognizable cluster of syndromes, loosely attributed to abnormal ciliary function and hence called ciliopathies. Regardless of the actual genetic cause, it is clustering of a set of characteristic physiological features which define whether a syndrome is a ciliopathy.
Although ciliopathies are usually considered to involve proteins that localize to motile and/or immotile (primary) cilia or centrosomes, it is possible for ciliopathies to be associated with unexpected proteins such as XPNPEP3, which localizes to mitochondria but is believed to affect ciliary function through proteolytic cleavage of ciliary proteins.[4]
Significant advances in understanding the importance of cilia were made in the mid-1990s. However, the physiological role that this organelle plays in most tissues remains elusive. Additional studies of how ciliary dysfunction can lead to such severe disease and developmental pathologies is still a subject of current research.[5]
Signs and symptoms
A wide variety of symptoms are potential clinical features of ciliopathy. The signs most exclusive to a ciliopathy, in descending order of exclusivity, are:[6]: 138
- Dandy–Walker malformation (cerebellar vermis hypoplasia, usually with hydrocephalus)
- Agenesis of the corpus callosum
- Situs inversus
- Posterior encephalocele
- Polycystic kidneys
- Postaxial polydactyly
- Liver disease
- Retinitis pigmentosa
- Intellectual disability
A case with polycystic ovary syndrome, multiple subcutaneous cysts, renal function impairment, Caroli disease and liver cirrhosis due to ciliopathy has been described.[7]
Phenotypes sometimes associated with ciliopathies can include:[6]
- Anencephaly
- Breathing abnormalities
- Cerebellar vermis hypoplasia
- Diabetes
- Exencephaly
- Eye movement abnormalities
- Hydrocephalus
- Hypoplasia of the corpus callosum
- Hypotonia
- Infertility
- Cognitive impairment/defects
- Obesity[8]
- Other polydactyly
- Respiratory dysfunction
- Renal cystic disease
- Retinal degeneration
- Sensorineural deafness
- Spina bifida
Pathophysiology
"In effect, the [motile cilium] is a nanomachine composed of perhaps over 600 proteins in molecular complexes, many of which also function independently as nanomachines." Cilia "function as mechano- or chemosensors and as a cellular global positioning system to detect changes in the surrounding environment." For example, ciliary signaling plays a role in the initiation of cellular replacement after cell damage.[9]
In addition to this sensory role mediating specific signaling cues, cilia play "a secretory role in which a soluble protein is released to have an effect downstream of the fluid flow" in epithelial cells, and can of course mediate fluid flow directly in the case of motile cilia.[1] Primary cilia in the retina play a role in transferring nourishment to the non-vascularized rod and cone cells from the vascularized cells several micrometres behind the surface of the retina.
Signal transduction pathways involved include the Hedgehog signaling pathway and the Wnt signaling pathway.[10]
Dysfunctional cilia can lead to:
- Chemosensation abnormalities,[11] typically via ciliated epithelial cellular dysfunction.[1]
- Defective thermosensation or mechanosensation,[12] often via ciliated epithelial cellular dysfunction.[1]
- Cellular motility dysfunction[11]
- Issues with displacement of extracellular fluid[11]
- Paracrine signal transduction abnormalities[1][11]
In organisms of normal health, cilia are critical for:[13]
- development
- homeostasis
- reproduction
Genetics
"Just as different genes can contribute to similar diseases, so the same genes and families of genes can play a part in a range of different diseases." For example, in just two of the diseases caused by malfunctioning cilia, Meckel–Gruber syndrome and Bardet–Biedl syndrome, patients who carry mutations in genes associated with both diseases "have unique symptoms that are not seen in either condition alone." The genes linked to the two different conditions "interact with each other during development." Systems biologists are endeavoring to define functional modules containing multiple genes and then look at disorders whose phenotypes fit into such modules.[14]
A particular phenotype can overlap "considerably with several conditions (ciliopathies) in which primary cilia are also implicated in pathogenicity. One emerging aspect is the wide spectrum of ciliopathy gene mutations found within different diseases."[8]
List of ciliopathies
"The phenotypic parameters that define a ciliopathy may be used to both recognize the cellular basis of a number of genetic disorders and to facilitate the diagnosis and treatment of some diseases of unknown" cause.[6]
Known ciliopathies
| Condition | OMIM | Gene(s) | Systems/organs affected |
|---|---|---|---|
| Alström syndrome[6][1] | 203800 | ALMS1 | |
| Asphyxiating thoracic dysplasia (Jeune syndrome)[6][15] | 208500 | ||
| Bardet–Biedl syndrome[6][5][8] | 209900 | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12 | |
| Ellis–van Creveld syndrome[15] | 225500 | EVC, EVC2 | |
| Joubert syndrome[6][8] | 213300 | INPP5E, TMEM216, AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, BRCC3 | Brain |
| Leber congenital amaurosis[15] | 204000 | GUCY2D, RPE65 | |
| McKusick–Kaufman syndrome[15] | 236700 | MKKS | |
| Meckel–Gruber syndrome[6][8][16] | 249000 | MKS1, TMEM67, TMEM216, CEP290, RPGRIP1L, CC2D2A | Liver, heart, bone |
| Nephronophthisis[6][5][8] | 256100 | NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, GLIS2, RPGRIP1L | Kidney |
| Orofaciodigital syndrome 1[1][5] | 311200 | OFD1 | |
| Polycystic kidney disease[6][5] (ADPKD and ARPKD)[17] | 173900 | PKD1, PKD2, PKHD1 | Kidney |
| Primary ciliary dyskinesia (Kartagener syndrome)[6] | 244400 | DNAI1, DNAH5, TXNDC3, DNAH11, DNAI2, KTU, RSPH4A, RSPH9, LRRC50 | |
| Senior–Løken syndrome[5] | 266900 | NPHP1, NPHP4, IQCB1, CEP290, SDCCAG8 | Eye |
| Sensenbrenner syndrome (cranioectodermal dysplasia)[15] | 218330 | IFT122 | |
| Short rib–polydactyly syndrome[15] | 613091 | DYNC2H1 | |
| ? | ? | IFT88 | Novel form of congenital anosmia, reported in 2012[18] |
Likely ciliopathies
| Condition | OMIM | Gene(s) | Systems/organs affected |
|---|---|---|---|
| Acrocallosal syndrome[15] | 200990 | KIF7, GLI3 | |
| Acromelic frontonasal dysostosis[15] | 603671 | ZSWIM6 | |
| Arima syndrome[15] | 243910 | ||
| Biemond syndrome[15] | 113400 | ||
| COACH syndrome[15] | 216360 | TMEM67, CC2D2A, RPGRIP1L | |
| Conorenal syndrome[19][15] | 266920 | ||
| Greig cephalopolysyndactyly syndrome[15] | 175700 | GLI3 | |
| Hydrolethalus syndrome[15] | 236680 | HYLS1 | |
| Johanson–Blizzard syndrome[15] | 243800 | UBR1 | |
| Mohr syndrome (oral-facial-digital syndrome type 2)[15] | 252100 | ||
| Neu–Laxova syndrome[15] | 256520 | PHGDH, PSAT1, PSPH | |
| Opitz G/BBB syndrome[15] | 300000 | MID1 | |
| Pallister–Hall syndrome[15] | 146510 | GLI3 | |
| Papillorenal syndrome[15] | 120330 | PAX2 | |
| Renal–hepatic–pancreatic dysplasia[15] | 208540 | NPHP3 | |
| Varadi–Papp syndrome (oral-facial-digital syndrome type 6)[15] | 277170 |
Possible ciliopathies
History
Although non-motile or primary cilia were first described in 1898, they were largely ignored by biologists. However, microscopists continued to document their presence in the cells of most vertebrate organisms. The primary cilium was long considered—with few exceptions—to be a largely useless evolutionary vestige, a vestigial organelle. Recent research has revealed that cilia are essential to many of the body's organs.[22] These primary cilia play important roles in chemosensation, mechanosensation, and thermosensation. Cilia may thus be "viewed as sensory cellular antennae that coordinate a large number of cellular signaling pathways, sometimes coupling the signaling to ciliary motility or alternatively to cell division and differentiation."[9]
Recent advances in mammalian genetic research have made possible the understanding of a molecular basis for a number of dysfunctional mechanisms in both motile and primary cilia structures of the cell.[23] A number of critical developmental signaling pathways essential to cellular development have been discovered. These are principally but not exclusively found in the non-motile or primary cilia. A number of common observable characteristics of mammalian genetic disorders and diseases are caused by ciliary dysgenesis and dysfunction. Once identified, these characteristics thus describe a set of hallmarks of a ciliopathy.[6]
Cilia have recently been implicated in a wide variety of human genetic diseases by "the discovery that numerous proteins involved in mammalian disease localize to the basal bodies and cilia." For example, in just a single area of human disease physiology, cystic renal disease, cilia-related genes and proteins have been identified to have causal effect in polycystic kidney disease, nephronophthisis, Senior–Løken syndrome type 5, orofaciodigital syndrome type 1 and Bardet–Biedl syndrome.[5]
References
- 1 2 3 4 5 6 7 Adams, M.; Smith, U. M.; Logan, C. V.; Johnson, C. A. (2008). "Recent advances in the molecular pathology, cell biology and genetics of ciliopathies". Journal of Medical Genetics. 45 (5): 257–267. doi:10.1136/jmg.2007.054999. PMID 18178628.
- ↑ Lee JH, Gleeson JG (May 2010). "The role of primary cilia in neuronal function". Neurobiol. Dis. 38 (2): 167–72. doi:10.1016/j.nbd.2009.12.022. PMC 2953617. PMID 20097287.
- ↑ Powles-Glover, N (September 2014). "Cilia and ciliopathies: classic examples linking phenotype and genotype-an overview". Reproductive Toxicology (Elmsford, N.Y.). 48: 98–105. doi:10.1016/j.reprotox.2014.05.005. PMID 24859270.
- ↑ Hurd TW, Hildebrandt F (2011). "Mechanisms of Nephronophthisis and Related Ciliopathies". Nephron Exp. Nephrol. 118 (1): e9–e14. doi:10.1159/000320888. PMC 2992643. PMID 21071979.
- 1 2 3 4 5 6 7 Davenport, J. R. (2005). "An incredible decade for the primary cilium: A look at a once-forgotten organelle". AJP: Renal Physiology. 289 (6): F1159–F1169. doi:10.1152/ajprenal.00118.2005. PMID 16275743.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Badano JL, Mitsuma N, Beales PL, Katsanis N (2006). "The ciliopathies: an emerging class of human genetic disorders". Annu Rev Genom Hum Genet. 7: 125–48. doi:10.1146/annurev.genom.7.080505.115610. PMID 16722803.
- ↑ Tan K, Liu P, Pang L, Yang W, Hou F (2018) A human ciliopathy with polycystic ovarian syndrome and multiple subcutaneous cysts: A rare case report. Medicine (Baltimore) 97(50)
- 1 2 3 4 5 6 Ross, Allison; PL Beales; J Hill (2008). The Clinical, Molecular, and Functional Genetics of Bardet-Biedl Syndrome, in Genetics of Obesity Syndromes. Oxford University Press. p. 177. ISBN 978-0-19-530016-1. Retrieved 2009-07-01.
- 1 2 Satir, Peter; Søren T. Christensen (2008-03-26). "Structure and function of mammalian cilia". Histochemistry and Cell Biology. Springer Berlin / Heidelberg. 129 (6): 687–693. doi:10.1007/s00418-008-0416-9. PMC 2386530. PMID 18365235. 1432-119X.
- ↑ D'Angelo A, Franco B (2009). "The dynamic cilium in human diseases". Pathogenetics. 2 (1): 3. doi:10.1186/1755-8417-2-3. PMC 2694804. PMID 19439065.
- 1 2 3 4 "Ciliary proteome database, v3". Database introduction. Johns Hopkins University. 2008. Retrieved 2009-01-07.
- ↑ Tan PL, Barr T, Inglis PN, et al. (2007). "Loss of Bardet–Biedl syndrome proteins causes defects in peripheral sensory innervation and function". Proc. Natl. Acad. Sci. U.S.A. 104 (44): 17524–9. doi:10.1073/pnas.0706618104. PMC 2077289. PMID 17959775.
- ↑ of organs The Ciliary Proteome, Ciliaproteome V3.0 - Home Page, accessed 2010-06-11.
- ↑ Hayden EC (2008). "Biological tools revamp disease classification". Nature. 453 (7196): 709. doi:10.1038/453709a. PMID 18528360.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 Baker, Kate; Beales, Philip L. (2009). "Making sense of cilia in disease: The human ciliopathies". American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 151C (4): 281–295. doi:10.1002/ajmg.c.30231. ISSN 1552-4876. PMID 19876933.
- ↑ Kyttälä, Mira (May 2006). "Identification of the Meckel Syndrome Gene (MKS1) Exposes a Novel Ciliopathy" (PDF). National Public Health Institute, Helsinki. Archived from the original (PDF) on 2006-07-21. Retrieved 2008-07-06.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Gunay-Aygun M (November 2009). "Liver and Kidney Disease in Ciliopathies". Am J Med Genet C Semin Med Genet. 151C (4): 296–306. doi:10.1002/ajmg.c.30225. PMC 2919058. PMID 19876928.
- ↑ Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model
- ↑ Watnick T, Germino G (August 2003). "From cilia to cyst". Nat. Genet. 34 (4): 355–6. doi:10.1038/ng0803-355. PMID 12923538.
- ↑ Delgado-Escueta AV (2007). "Advances in Genetics of Juvenile Myoclonic Epilepsies". Epilepsy Curr. 7 (3): 61–7. doi:10.1111/j.1535-7511.2007.00171.x. PMC 1874323. PMID 17520076.
- ↑ Khanna, H.; Davis, E. E.; Murga-Zamalloa, C. A.; et al. (2009). "A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies". Nature Genetics. 41 (6): 739–745. doi:10.1038/ng.366. PMC 2783476. PMID 19430481.
- ↑ Gardiner, Mary Beth (September 2005). "The Importance of Being Cilia". HHMI Bulletin. Howard Hughes Medical Institute. 18 (2). Archived from the original on 2010-03-11. Retrieved 2008-07-26.
- ↑ Lancaster MA, Gleeson JG (June 2009). "The primary cilium as a cellular signaling center: lessons from disease". Curr. Opin. Genet. Dev. 19 (3): 220–9. doi:10.1016/j.gde.2009.04.008. PMC 2953615. PMID 19477114.