X-linked hypophosphatemia
| X-linked hypophosphatemia | |
|---|---|
| Other names: X-linked dominant hypophosphatemic rickets, or X-linked Vitamin D-resistant rickets,[1] | |
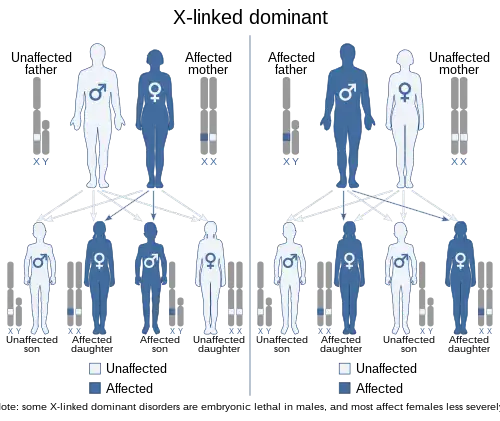 | |
| This condition is inherited in an X-linked dominant manner. | |
| Complications | osteomalacia (adults), rickets (children), fractures, enthesopathy, spinal stenosis, abnormal gait, short stature, tinnitus, hearing loss, dental complications, in rare exceptions Chiari malformation can occur. |
| Causes | A genetic mutation of the PHEX gene results in elevated FGF23 hormone. |
| Medication | phosphate, vitamin-D or burosumab |
X-linked hypophosphatemia (XLH), (or Hypophosphatemic rickets) is an X-linked dominant form of rickets (or osteomalacia) that differs from most cases of rickets in that vitamin D supplementation does not cure it. It can cause bone deformity including short stature and genu varum (bow-leggedness). It is associated with a mutation in the PHEX gene sequence (Xp.22) and subsequent inactivity of the PHEX protein.[2] The prevalence of the disease is 1 in 20,000.[3]
X-linked hypophosphatemia may be lumped in with Autosomal dominant hypophosphatemic rickets under general terms such as hypophosphatemic rickets. Hypophospatemic rickets are associated with at least nine other genetic mutations.[4] Clinical management of hypophospatemic rickets may differ depending on the specific mutations associated with an individual case, but treatments are aimed at raising phosphate levels to promote normal bone formation.[5]
Symptoms and signs
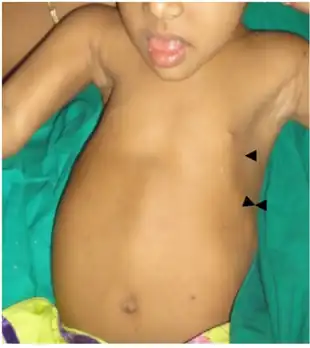
The most common symptoms of XLH affect the bones and teeth, causing pain, abnormalities, and osteoarthritis. Symptoms and signs can vary between children and adults and can include:
Children
- Rickets
- Dental abscesses
- Craniostenosis
- Fractures / pseudofractures
- Bone pain
- Fatigue
- Delayed and disproportionate growth
- Delayed motor development and gait abnormalities
Adults
Genetics
XLH is associated with a mutation in the PHEX gene sequence, located on the human X chromosome at location Xp22.2-p22.1.[1][2][9] The PHEX protein regulates another protein called fibroblast growth factor 23 (produced from the FGF23 gene). Fibroblast growth factor 23 normally inhibits the kidneys' ability to reabsorb phosphate into the bloodstream. Gene mutations in PHEX prevent it from correctly regulating fibroblast growth factor 23. The resulting overactivity of FGF-23 reduces vitamin D 1α-hydroxylation and phosphate reabsorption by the kidneys, leading to hypophosphatemia and the related features of hereditary hypophosphatemic rickets.[10] Also in XLH, where PHEX enzymatic activity is absent or reduced, osteopontin[11]—a mineralization-inhibiting secreted substrate protein found in the extracellular matrix of bone[12]—accumulates in bone (and teeth) to contribute to the osteomalacia (and odontomalacia) as shown in the mouse homolog (Hyp) of XLH and in XLH patients.[13][14][15] Biochemically in blood, XLH is recognized by hypophosphatemia and an inappropriately low level of calcitriol (1,25-(OH)2 vitamin D3). Patients often have bowed legs or knock knees in which they usually cannot touch both knees and ankles together at the same time.
The disorder is inherited in an X-linked dominant manner.[1][2] This means the defective gene responsible for the disorder (PHEX) is located on the X chromosome, and only one copy of the defective gene is sufficient to cause the disorder when inherited from a parent who has the disorder. Males are normally hemizygous for the X chromosome, having only one copy. As a result, X-linked dominant disorders usually show higher expressivity in males than females.
As the X chromosome is one of the sex chromosomes (the other being the Y chromosome), X-linked inheritance is determined by the sex of the parent carrying a specific gene and can often seem complex. This is because, typically, females have two copies of the X-chromosome and males have only one copy. The difference between dominant and recessive inheritance patterns also plays a role in determining the chances of a child inheriting an X-linked disorder from their parentage.
Diagnosis
Begin clinical laboratory evaluation of rickets with assessment of serum calcium, phosphate, and alkaline phosphatase levels. In hypophosphatemic rickets, calcium levels may be within or slightly below the reference range; alkaline phosphatase levels will be significantly above the reference range.
Carefully evaluate serum phosphate levels in the first year of life, because the concentration reference range for infants (5.0-7.5 mg/dL) is high compared with that for adults (2.7-4.5 mg/dL).
Serum parathyroid hormone levels are within the reference range or slightly elevated, while calcitriol levels are low or within the lower reference range. Most importantly, urinary loss of phosphate is above the reference range.
The renal tubular reabsorption of phosphate (TRP) in X-linked hypophosphatemia is 60%; normal TRP exceeds 90% at the same reduced plasma phosphate concentration. The TRP is calculated with the following formula:
1 - [Phosphate Clearance (CPi) / Creatinine Clearance (Ccr)] X 100
Treatment
Oral phosphate,[16][17] calcitriol;[16][17] in the event of severe bowing, an osteotomy may be performed to correct the leg shape.[18] The monoclonal antibody Burosumab was first licensed in February 2018 by the European Medicines Agency,[19] then licensed by the Food and Drug Administration in the United States of America in June 2018 [20]as the first drug targeting the underlying cause for this condition.[21]
The leg deformity can be treated with Ilizarov frames and CAOS. It is also treated with medications including human growth hormone, calcitriol, and phosphate.[18]
Society and Culture
International XLH Alliance Archived 2021-03-21 at the Wayback Machine - an alliance of international patient groups for individuals affected by XLH and related disorders.
Jennyfer Marques Parinos is a Paralympic bronze medalist from Brazil who has XLH. She competes under a class 9 disability.
See also
References
- 1 2 3 Online Mendelian Inheritance in Man (OMIM): 307800"HYPOPHOSPHATEMIC RICKETS, X-LINKED DOMINANT; XLHR". 23 May 2011.
{{cite web}}: Missing or empty|url=(help) - 1 2 3 Saito, T.; Nishii, Y.; Yasuda, T.; Ito, N.; Suzuki, H.; Igarashi, T.; Fukumoto, S.; Fujita, T. (Oct 2009). "Familial hypophosphatemic rickets caused by a large deletion in PHEX gene". European Journal of Endocrinology. 161 (4): 647–651. doi:10.1530/EJE-09-0261. PMID 19581284.
- ↑ Carpenter TO (Apr 1997). "New perspectives on the biology and treatment of X-linked hypophosphatemic rickets". Pediatr. Clin. North Am. 44 (2): 443–466. doi:10.1016/S0031-3955(05)70485-5. PMID 9130929.
- ↑ Online Mendelian Inheritance in Man (OMIM): 193100
- ↑ "Hypophosphatemic rickets". Genetic and Rare Diseases Information Center. National Institutes of Health. Archived from the original on 10 October 2012. Retrieved 10 October 2012.
- 1 2 3 4 5 Linglart, A.; Biosse-Duplan, M.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; Wicart, P.; Harvengt, P. (2014). "Therapeutic management of hypophosphatemic rickets from infancy to adulthood". Endocrine Connections. 3 (1): R13–R30. doi:10.1530/EC-13-0103. PMC 3959730. PMID 24550322.
- ↑ Skrinar, A.; Dvorak-Ewell, M.; Evins, A.; MacIca, C.; Linglart, A.; Imel, E. A.; Theodore-Oklota, C.; San Martin, J. (2019). "The Lifelong Impact of X-Linked Hypophosphatemia: Results From a Burden of Disease Survey". Journal of the Endocrine Society. 3 (7): 1321–1334. doi:10.1210/js.2018-00365. PMC 6595532. PMID 31259293.
- ↑ Hawley, S.; Shaw, N. J.; Delmestri, A.; Prieto-Alhambra, D.; Cooper, C.; Pinedo-Villanueva, R.; Javaid, M. K. (2020). "Higher prevalence of non-skeletal comorbidity related to X-linked hypophosphataemia: a UK parallel cohort study using CPRD". Rheumatology. doi:10.1093/rheumatology/keaa859. PMID 33331900.
- ↑ 300550"PHOSPHATE-REGULATING ENDOPEPTIDASE HOMOLOG, X-LINKED; PHEX". 18 April 2011.
{{cite web}}: Missing or empty|url=(help) - ↑ Perwad, Farzana; Zhang, Martin Y. H.; Tenenhouse, Harriet S.; Portale, Anthony A. (2007-11-01). "Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro". American Journal of Physiology. Renal Physiology. 293 (5): F1577–1583. doi:10.1152/ajprenal.00463.2006. ISSN 1931-857X. PMID 17699549.
- ↑ Sodek, J; et al. (2000). "Osteopontin". Critical Reviews in Oral Biology and Medicine. 11 (3): 279–303. doi:10.1177/10454411000110030101. PMID 11021631.
- ↑ McKee, MD; et al. (2005). "Hierarchies of extracellular matrix and mineral organization in bone of the craniofacial complex and skeleton". Cells Tissues Organs. 181 (3–4): 176–188. doi:10.1159/000091379. PMID 16612083. S2CID 40705942. Archived from the original on 2021-08-27. Retrieved 2021-08-14.
- ↑ McKee, MD; Hoac, B; Addison, WN; Barros, NM; Millán, JL; Chaussain, C (October 2013). "Extracellular matrix mineralization in periodontal tissues: Noncollagenous matrix proteins, enzymes, and relationship to hypophosphatasia and X-linked hypophosphatemia". Periodontology 2000. 63 (1): 102–22. doi:10.1111/prd.12029. PMC 3766584. PMID 23931057.
- ↑ Boukpessi, T; Hoac, B; Coyac, BR; Leger, T; Garcia, C; Wicart, P; Whyte, MP; Glorieux, FH; Linglart, A; Chaussain, C; McKee, MD (21 November 2016). "Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia". Bone. 95: 151–161. doi:10.1016/j.bone.2016.11.019. PMID 27884786.
- ↑ Barros, NMT; et al. (2013). "Proteolytic processing of osteopontin by PHEX and accumulation of osteopontin fragments in Hyp mouse bone, the murine model of X-linked hypophosphatemia". Journal of Bone and Mineral Research. 28 (3): 688–699. doi:10.1002/jbmr.1766. PMID 22991293.
- 1 2 Imel, E. A.; DiMeglio, L. A.; Hui, S. L.; Carpenter, T. O.; Econs, M. J. (15 February 2010). "Treatment of X-Linked Hypophosphatemia with Calcitriol and Phosphate Increases Circulating Fibroblast Growth Factor 23 Concentrations". Journal of Clinical Endocrinology & Metabolism. 95 (4): 1846–1850. doi:10.1210/jc.2009-1671. PMC 2853995. PMID 20157195.
- 1 2 Glorieux, F. H.; Marie, P. J.; Pettifor, J. M.; Delvin, E. E. (30 October 1980). "Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets". The New England Journal of Medicine. 303 (18): 1023–1031. doi:10.1056/NEJM198010303031802. PMID 6252463.
- 1 2 "X-linked hypophosphatemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2019-04-03. Retrieved 2018-10-21.
- ↑ "EMA authorisation details". Archived from the original on 2021-07-25.
- ↑ "FDA press release". Archived from the original on 2021-07-26.
- ↑ Carpenter, TO; Whyte MP; Imel EA; Boot AM; Högler W; Linglart A; Padidela R; Van't Hoff W; Mao M; Chen CY; Skrinar A; Kakkis E; San Martin J; Portale AA (24 May 2018). "Burosumab Therapy in Children with X-Linked Hypophosphatemia". The New England Journal of Medicine (Submitted manuscript). 378 (21): 1987–1998. doi:10.1056/NEJMoa1714641. hdl:1805/18603. PMID 29791829. S2CID 44135503. Archived from the original on 3 April 2019. Retrieved 14 August 2021.
External links
- Hypophosphatemic rickets; XLH; Hypophosphatemia, vitamin D-resistant rickets at NIH's Office of Rare Diseases
- The PHEXdb Archived 2012-04-01 at the Wayback Machine - a database of nucleotide variation in the PHEX gene
| Classification | |
|---|---|
| External resources |
|