Retinoblastoma protein
The retinoblastoma protein (protein name abbreviated Rb; gene name abbreviated Rb, RB or RB1) is a tumor suppressor protein that is dysfunctional in several major cancers.[5] One function of pRb is to prevent excessive cell growth by inhibiting cell cycle progression until a cell is ready to divide. When the cell is ready to divide, pRb is phosphorylated, inactivating it, and the cell cycle is allowed to progress. It is also a recruiter of several chromatin remodeling enzymes such as methylases and acetylases.[6]




pRb belongs to the pocket protein family, whose members have a pocket for the functional binding of other proteins.[7][8] Should an oncogenic protein, such as those produced by cells infected by high-risk types of human papillomavirus, bind and inactivate pRb, this can lead to cancer. The RB gene may have been responsible for the evolution of multicellularity in several lineages of life including animals.[9]
Name and genetics
In humans, the protein is encoded by the RB1 gene located on chromosome 13—more specifically, 13q14.1-q14.2. If both alleles of this gene are mutated in a retinal cell, the protein is inactivated and the cells grows uncontrollably, resulting in development of retinoblastoma cancer, hence the "RB" in the name 'pRb'. Thus most pRb knock-outs occur in retinal tissue when UV radiation-induced mutation inactivates all healthy copies of the gene, but pRb knock-out has also been documented in certain skin cancers in patients from New Zealand where the amount of UV radiation is significantly higher.
Two forms of retinoblastoma were noticed: a bilateral, familial form and a unilateral, sporadic form. Sufferers of the former were over six times more likely to develop other types of cancer later in life, compared to individuals with sporadic retinoblastoma.[10] This highlighted the fact that mutated pRb could be inherited and lent support for the two-hit hypothesis. This states that only one working allele of a tumour suppressor gene is necessary for its function (the mutated gene is recessive), and so both need to be mutated before the cancer phenotype will appear. In the familial form, a mutated allele is inherited along with a normal allele. In this case, should a cell sustain only one mutation in the other RB gene, all pRb in that cell would be ineffective at inhibiting cell cycle progression, allowing cells to divide uncontrollably and eventually become cancerous. Furthermore, as one allele is already mutated in all other somatic cells, the future incidence of cancers in these individuals is observed with linear kinetics.[11] The working allele need not undergo a mutation per se, as loss of heterozygosity (LOH) is frequently observed in such tumours.
However, in the sporadic form, both alleles would need to sustain a mutation before the cell can become cancerous. This explains why sufferers of sporadic retinoblastoma are not at increased risk of cancers later in life, as both alleles are functional in all their other cells. Future cancer incidence in sporadic pRb cases is observed with polynomial kinetics, not exactly quadratic as expected because the first mutation must arise through normal mechanisms, and then can be duplicated by LOH to result in a tumour progenitor.
RB1 orthologs[12] have also been identified in most mammals for which complete genome data are available.
RB/E2F-family proteins repress transcription.[13]
Structure denotes function
pRb is a multifunctional protein with many binding and phosphorylation sites. Although its common function is seen as binding and repressing E2F targets, pRb is likely a multifunctional protein as it binds to at least 100 other proteins.[14]
pRb has three major structural components: a carboxy-terminus, a "pocket" subunit, and an amino-terminus. Within each domain, there are a variety of protein binding sites, as well as a total of 15 possible phosphorylation sites. Generally, phosphorylation causes interdomain locking, which changes pRb's conformation and prevents binding to target proteins. Different sites may be phosphorylated at different times, giving rise to many possible conformations and likely many functions/activity levels.[15]
Cell cycle suppression

pRb restricts the cell's ability to replicate DNA by preventing its progression from the G1 (first gap phase) to S (synthesis phase) phase of the cell division cycle.[16] pRb binds and inhibits E2 promoter-binding–protein-dimerization partner (E2F-DP) dimers, which are transcription factors of the E2F family that push the cell into S phase.[17][18][19][20][21][22] By keeping E2F-DP inactivated, RB1 maintains the cell in the G1 phase, preventing progression through the cell cycle and acting as a growth suppressor.[8] The pRb-E2F/DP complex also attracts a histone deacetylase (HDAC) protein to the chromatin, reducing transcription of S phase promoting factors, further suppressing DNA synthesis.
pRb attenuates protein levels of known E2F Targets
pRb has the ability to reversibly inhibit DNA replication through transcriptional repression of DNA replication factors. pRb is able to bind to transcription factors in the E2F family and thereby inhibit their function. When pRb is chronically activated, it leads to the downregulation of the necessary DNA replication factors. Within 72–96 hours of active pRb induction in A2-4 cells, the target DNA replication factor proteins—MCMs, RPA34, DBF4, RFCp37, and RFCp140—all showed decreased levels. Along with decreased levels, there was a simultaneous and expected inhibition of DNA replication in these cells. This process, however, is reversible. Following induced knockout of pRb, cells treated with cisplatin, a DNA-damaging agent, were able to continue proliferating, without cell cycle arrest, suggesting pRb plays an important role in triggering chronic S-phase arrest in response to genotoxic stress.
One such example of E2F-regulated genes repressed by pRb are cyclin E and cyclin A. Both of these cyclins are able to bind to Cdk2 and facilitate entry into the S phase of the cell cycle. Through the repression of expression of cyclin E and cyclin A, pRb is able to inhibit the G1/S transition.
Repression mechanisms of E2Fs
There are at least three distinct mechanisms in which pRb can repress transcription of E2F-regulated promoters. Though these mechanisms are known, it is unclear which are the most important for the control of the cell cycle.
E2Fs are a family of proteins whose binding sites are often found in the promoter regions of genes for cell proliferation or progression of the cell cycle. E2F1 to E2F5 are known to associate with proteins in the pRb-family of proteins while E2F6 and E2F7 are independent of pRb. Broadly, the E2Fs are split into activator E2Fs and repressor E2Fs though their role is more flexible than that on occasion. The activator E2Fs are E2F1, E2F2 and E2F3 while the repressor E2Fs are E2F4, E2F5 and E2F6. Activator E2Fs along with E2F4 bind exclusively to pRb. pRb is able to bind to the activation domain of the activator E2Fs which blocks their activity, repressing transcription of the genes controlled by that E2F-promoter.
Blocking of pre-initiation complex assembly
The preinitiation complex (PIC) assembles in a stepwise fashion on the promoter of genes to initiate transcription. The TFIID binds to the TATA box in order to begin the assembly of the TFIIA, recruiting other transcription factors and components needed in the PIC. Data suggests that pRb is able to repress transcription by both pRb being recruited to the promoter as well as having a target present in TFIID.
The presence of pRb may change the conformation of the TFIIA/IID complex into a less active version with a decreased binding affinity. pRb can also directly interfere with their association as proteins, preventing TFIIA/IID from forming an active complex.
Modification of chromatin structure
pRb acts as a recruiter that allows for the binding of proteins that alter chromatin structure onto the site E2F-regulated promoters. Access to these E2F-regulated promoters by transcriptional factors is blocked by the formation of nucleosomes and their further packing into chromatin. Nucleosome formation is regulated by post-translational modifications to histone tails. Acetylation leads to the disruption of nucleosome structure. Proteins called histone acetyltransferases (HATs) are responsible for acetylating histones and thus facilitating the association of transcription factors on DNA promoters. Deacetylation, on the other hand, leads to nucleosome formation and thus makes it more difficult for transcription factors to sit on promoters. Histone deacetylases (HDACs) are the proteins responsible for facilitating nucleosome formation and are therefore associated with transcriptional repressors proteins.
pRb interacts with the histone deacetylases HDAC1 and HDAC3. pRb binds to HDAC1 in its pocket domain in a region that is independent to its E2F-binding site. pRb recruitment of histone deacetylases leads to the repression of genes at E2F-regulated promoters due to nucleosome formation. Some genes activated during the G1/S transition such as cyclin E are repressed by HDAC during early to mid-G1 phase. This suggests that HDAC-assisted repression of cell cycle progression genes is crucial for the ability of pRb to arrest cells in G1. To further add to this point, the HDAC-pRb complex is shown to be disrupted by cyclin D/Cdk4 which levels increase and peak during the late G1 phase.
Senescence induced by pRb
Senescence in cells is a state in which cells are metabolically active but are no longer able to replicate. pRb is an important regulator of senescence in cells and since this prevents proliferation, senescence is an important antitumor mechanism. pRb may occupy E2F-regulated promoters during senescence. For example, pRb was detected on the cyclin A and PCNA promoters in senescent cells.
S-phase arrest
Cells respond to stress in the form of DNA damage, activated oncogenes, or sub-par growing conditions, and can enter a senescence-like state called "premature senescence". This allows the cell to prevent further replication during periods of damaged DNA or general unfavorable conditions. DNA damage in a cell can induce pRb activation. pRb's role in repressing the transcription of cell cycle progression genes leads to the S phase arrest that prevents replication of damaged DNA.
Activation and inactivation
When it is time for a cell to enter S phase, complexes of cyclin-dependent kinases (CDK) and cyclins phosphorylate pRb, allowing E2F-DP to dissociate from pRb and become active.[8] When E2F is free it activates factors like cyclins (e.g. cyclin E and cyclin A), which push the cell through the cell cycle by activating cyclin-dependent kinases, and a molecule called proliferating cell nuclear antigen, or PCNA, which speeds DNA replication and repair by helping to attach polymerase to DNA.[18][21][7][8][19][23][24]
Inactivation
Since the 1990s, pRb was known to be inactivated via phosphorylation. Until, the prevailing model was that Cyclin D- Cdk 4/6 progressively phosphorylated it from its unphosphorylated to its hyperphosphorylated state (14+ phosphorylations). However, it was recently shown that pRb only exists in three states: un-phosphorylated, mono-phosphorylated, and hyper-phosphorylated. Each has a unique cellular function.[25]
Before the development of 2D IEF, only hyper-phosphorylated pRb was distinguishable from all other forms, i.e. un-phosphorylated pRb resembled mono-phosphorylated pRb on immunoblots. As pRb was either in its active “hypo-phosphorylated” state or inactive “hyperphosphorylated” state. However, with 2D IEF, it is now known that pRb is un-phosphorylated in G0 cells and mono-phosphorylated in early G1 cells, prior to hyper-phosphorylation after the restriction point in late G1.[25]
pRb mono phosphorylation
When a cell enters G1, Cyclin D- Cdk4/6 phosphorylates pRb at a single phosphorylation site. No progressive phosphorylation occurs because when HFF cells were exposed to sustained cyclin D- Cdk4/6 activity (and even deregulated activity) in early G1, only mono-phosphorylated pRb was detected. Furthermore, triple knockout, p16 addition, and Cdk 4/6 inhibitor addition experiments confirmed that Cyclin D- Cdk 4/6 is the sole phosphorylator of pRb.[25]
Throughout early G1, mono-phosphorylated pRb exists as 14 different isoforms (the 15th phosphorylation site is not conserved in primates in which the experiments were performed). Together, these isoforms represent the “hypo-phosphorylated” active pRb state that was thought to exist. Each isoform has distinct preferences to associate with different exogenous expressed E2Fs.[25]
A recent report showed that mono-phosphorylation controls pRb's association with other proteins and generates functional distinct forms of pRb.[26] All different mono-phosphorylated pRb isoforms inhibit E2F transcriptional program and are able to arrest cells in G1-phase. Importantly, different mono-phosphorylated forms of pRb have distinct transcriptional outputs that are extended beyond E2F regulation.[26]
Hyper-phosphorylation
After a cell passes the restriction point, Cyclin E - Cdk 2 hyper-phosphorylates all mono-phosphorylated isoforms. While the exact mechanism is unknown, one hypothesis is that binding to the C-terminus tail opens the pocket subunit, allowing access to all phosphorylation sites. This process is hysteretic and irreversible, and it is thought accumulation of mono-phosphorylated pRb induces the process. The bistable, switch like behavior of pRb can thus be modeled as a bifurcation point:[25]
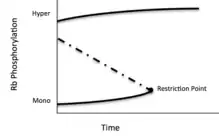
Control of pRb function by phosphorylation
Presence of un-phosphorylated pRb drives cell cycle exit and maintains senescence. At the end of mitosis, PP1 dephosphorylates hyper-phosphorylated pRb directly to its un-phosphorylated state. Furthermore, when cycling C2C12 myoblast cells differentiated (by being placed into a differentiation medium), only un-phosphorylated pRb was present. Additionally, these cells had a markedly decreased growth rate and concentration of DNA replication factors (suggesting G0 arrest).[25]
This function of un-phosphorylated pRb gives rise to a hypothesis for the lack of cell cycle control in cancerous cells: Deregulation of Cyclin D - Cdk 4/6 phosphorylates un-phosphorylated pRb in senescent cells to mono-phosphorylated pRb, causing them to enter G1. The mechanism of the switch for Cyclin E activation is not known, but one hypothesis is that it is a metabolic sensor. Mono-phosphorylated pRb induces an increase in metabolism, so the accumulation of mono-phosphorylated pRb in previously G0 cells then causes hyper-phosphorylation and mitotic entry. Since any un-phosphorylated pRb is immediately phosphorylated, the cell is then unable to exit the cell cycle, resulting in continuous division.[25]
DNA damage to G0 cells activates Cyclin D - Cdk 4/6, resulting in mono-phosphorylation of un-phosphorylated pRb. Then, active mono-phosphorylated pRb causes repression of E2F-targeted genes specifically. Therefore, mono-phosphorylated pRb is thought to play an active role in DNA damage response, so that E2F gene repression occurs until the damage is fixed and the cell can pass the restriction point. As a side note, the discovery that damages causes Cyclin D - Cdk 4/6 activation even in G0 cells should be kept in mind when patients are treated with both DNA damaging chemotherapy and Cyclin D - Cdk 4/6 inhibitors.[25]
Activation
During the M-to-G1 transition, pRb is then progressively dephosphorylated by PP1, returning to its growth-suppressive hypophosphorylated state.[8][27]
pRb family proteins are components of the DREAM complex composed of DP, E2F4/5, RB-like (p130/p107) And MuvB (Lin9:Lin37:Lin52:RbAbP4:Lin54). The DREAM complex is assembled in Go/G1 and maintains quiescence by assembling at the promoters of > 800 cell-cycle genes and mediating transcriptional repression. Assembly of DREAM requires DYRK1A (Ser/Thr kinase) dependant phosphorylation of the MuvB core component, Lin52 at Serine28. This mechanism is crucial for recruitment of p130/p107 to the MuvB core and thus DREAM assembly.
Consequences of pRb loss
Consequences of loss of pRb function is dependent on cell type and cell cycle status, as pRb's tumor suppressive role changes depending on the state and current identity of the cell.
In G0 quiescent stem cells, pRb is proposed to maintain G0 arrest although the mechanism remains largely unknown. Loss of pRb leads to exit from quiescence and an increase in the number of cells without loss of cell renewal capacity. In cycling progenitor cells, pRb plays a role at the G1, S, and G2 checkpoints and promotes differentiation. In differentiated cells, which make up the majority of cells in the body and are assumed to be in irreversible G0, pRb maintains both arrest and differentiation.[28]
Loss of pRb therefore exhibits multiple different responses within different cells that ultimately all could result in cancer phenotypes. For cancer initiation, loss of pRb may induce cell cycle re-entry in both quiescent and post-mitotic differentiated cells through dedifferentiation. In cancer progression, loss of pRb decreases the differentiating potential of cycling cells, increases chromosomal instability, prevents induction of cellular senescence, promotes angiogenesis, and increases metastatic potential.[28]
Although most cancers rely on glycolysis for energy production (Warburg effect),[29] cancers due to pRb loss tend to upregulate oxidative phosphorylation.[30] The increased oxidative phosphorylation can increase stemness, metastasis, and (when enough oxygen is available) cellular energy for anabolism.[30]
In vivo, it is still not entirely clear how and which cell types cancer initiation occurs with solely loss of pRb, but it is clear that the pRb pathway is altered in large number of human cancers.[110] In mice, loss of pRb is sufficient to initiate tumors of the pituitary and thyroid glands, and mechanisms of initiation for these hyperplasia are currently being investigated.[31]
Non-canonical roles
The classic view of pRb's role as a tumor suppressor and cell cycle regulator developed through research investigating mechanisms of interactions with E2F family member proteins. Yet, more data generated from biochemical experiments and clinical trials reveal other functions of pRb within the cell unrelated (or indirectly related) to tumor suppression.[32]
Functional hyperphosphorylated pRb
In proliferating cells, certain pRb conformations (when RxL motif if bound by protein phosphatase 1 or when it is acetylated or methylated) are resistant to CDK phosphorylation and retain other function throughout cell cycle progression, suggesting not all pRb in the cell are devoted to guarding the G1/S transition.[32]
Studies have also demonstrated that hyperphosphorylated pRb can specifically bind E2F1 and form stable complexes throughout the cell cycle to carry out unique unexplored functions, a surprising contrast from the classical view of pRb releasing E2F factors upon phosphorylation.[32]
In summary, many new findings about pRb's resistance to CDK phosphorylation are emerging in pRb research and shedding light on novel roles of pRb beyond cell cycle regulation.
Genome stability
pRb is able to be localize to sites of DNA breaks during the repair process and assist in non-homologous end joining and homologous recombination through complexing with E2F1. Once at the breaks, pRb is able to recruit regulators of chromatin structure such as the DNA helicase transcription activator BRG1. pRb has been shown to also be able to recruit protein complexes such as condensin and cohesin to assist in the structural maintenance of chromatin.[32]
Such findings suggest that in addition to its tumor suppressive role with E2F, pRb is also distributed throughout the genome to aid in important processes of genome maintenance such as DNA break-repair, DNA replication, chromosome condensation, and heterochromatin formation.[32]
Regulation of metabolism
pRb has also been implicated in regulating metabolism through interactions with components of cellular metabolic pathways. RB1 mutations can cause alterations in metabolism, including reduced mitochondrial respiration, reduced activity in the electron transport chain, and changes in flux of glucose and/or glutamine. Particular forms of pRb have been found to localize to the outer mitochondrial membrane and directly interacts with Bax to promote apoptosis.[33]
As a drug target
pRb Reactivation
While the frequency of alterations of the RB gene is substantial for many human cancer types including as lung, esophageal, and liver, alterations in up-steam regulatory components of pRb such as CDK4 and CDK6 have been the main targets for potential therapeutics to treat cancers with dysregulation in the RB pathway.[34] This focus has resulted in the recent development and FDA clinical approval of three small molecule CDK4/6 inhibitors (Palbociclib (IBRANCE, Pfizer Inc. 2015), Ribociclib (KISQUALI, Novartis. 2017), & Abemaciclib (VERZENIO, Eli Lilly. 2017)) for the treatment of specific breast cancer subtypes. However, recent clinical studies finding limited efficacy, high toxicity, and acquired resistance[35][36] of these inhibitors suggests the need to further elucidate mechanisms that influence CDK4/6 activity as well as explore other potential targets downstream in the pRb pathway to reactivate pRb's tumor suppressive functions. Treatment of cancers by CDK4/6 inhibitors depends on the presence of pRb within the cell for therapeutic effect, limiting their usage only to cancers where RB is not mutated and pRb protein levels are not significantly depleted.[34]
Direct pRb reactivation in humans has not been achieved. However, in murine models, novel genetic methods have allowed for in vivo pRb reactivation experiments. pRb loss induced in mice with oncogenic KRAS-driven tumors of lung adenocarcinoma negates the requirement of MAPK signal amplification for progression to carcinoma and promotes loss of lineage commitment as well as accelerate the acquisition of metastatic competency. Reactivation of pRb in these mice rescues the tumors towards a less metastatic state, but does not completely stop tumor growth due to a proposed rewiring of MAPK pathway signaling, which suppresses pRb through a CDK-dependent mechanism.[37]
Pro-apoptotic effects of pRb loss
Besides trying to re-activate the tumor suppressive function of pRb, one other distinct approach to treat dysregulated pRb pathway cancers is to take advantage of certain cellular consequences induced by pRb loss. It has been shown that E2F stimulates expression of pro-apoptotic genes in addition to G1/S transition genes, however, cancer cells have developed defensive signaling pathways that protect themselves from death by deregulated E2F activity. Development of inhibitors of these protective pathways could thus be a synthetically lethal method to kill cancer cells with overactive E2F.[34]
In addition, it has been shown that the pro-apoptotic activity of p53 is restrained by the pRb pathway, such that pRb deficient tumor cells become sensitive to p53 mediated cell death. This opens the door to research of compounds that could activate p53 activity in these cancer cells and induce apoptosis and reduce cell proliferation.[34]
Regeneration
While the loss of a tumor suppressor such as pRb leading to uncontrolled cell proliferation is detrimental in the context of cancer, it may be beneficial to deplete or inhibit suppressive functions of pRb in the context of cellular regeneration.[38] Harvesting the proliferative abilities of cells induced to a controlled “cancer like” state could aid in repairing damaged tissues and delay aging phenotypes. This idea remains to be thoroughly explored as a potential cellular injury and anti-aging treatment.
Cochlea
The retinoblastoma protein is involved in the growth and development of mammalian hair cells of the cochlea, and appears to be related to the cells' inability to regenerate. Embryonic hair cells require pRb, among other important proteins, to exit the cell-cycle and stop dividing, which allows maturation of the auditory system. Once wild-type mammals have reached adulthood, their cochlear hair cells become incapable of proliferation. In studies where the gene for pRb is deleted in mice cochlea, hair cells continue to proliferate in early adulthood. Though this may seem to be a positive development, pRb-knockdown mice tend to develop severe hearing loss due to degeneration of the organ of Corti. For this reason, pRb seems to be instrumental for completing the development of mammalian hair cells and keeping them alive.[39][40] However, it is clear that without pRb, hair cells have the ability to proliferate, which is why pRb is known as a tumor suppressor. Temporarily and precisely turning off pRb in adult mammals with damaged hair cells may lead to propagation and therefore successful regeneration. Suppressing function of the retinoblastoma protein in the adult rat cochlea has been found to cause proliferation of supporting cells and hair cells. pRb can be downregulated by activating the sonic hedgehog pathway, which phosphorylates the proteins and reduces gene transcription.[41]
Neurons
Disrupting pRb expression in vitro, either by gene deletion or knockdown of pRb short interfering RNA, causes dendrites to branch out farther. In addition, Schwann cells, which provide essential support for the survival of neurons, travel with the neurites, extending farther than normal. The inhibition of pRb supports the continued growth of nerve cells.[42]
Interactions
pRb is known to interact with more than 300 proteins, some of which are listed below:
- Abl gene[43][44]
- Androgen receptor[45][46]
- Apoptosis-antagonizing transcription factor[47][48]
- ARID4A[49]
- Aryl hydrocarbon receptor[50]
- BRCA1[51][52][53]
- BRF1[54][55]
- C-jun[56]
- C-Raf[57][58]
- CDK9[59]
- CUTL1[60]
- Cyclin A1[61]
- Cyclin D1[62][63]
- Cyclin T2[59]
- DNMT1[64]
- E2F1[65][66][67][68][69][17][70]
- E2F2,[71]
- E4F1[68]
- EID1[72][73]
- ENC1[74]
- FRK[75]
- HBP1[76]
- HDAC1[49][77][78][79][80][81][82]
- HDAC3[49][83]
- Histone deacetylase 2[49]
- Insulin[84]
- JARID1A[85][86]
- Large tumor antigen[87][88]
- LIN9[89]
- MCM7[90]
- MORF4L1[66][91]
- MRFAP1,[66][91]
- MyoD[92][93]
- NCOA6[94]
- PA2G4[95]
- Peroxisome proliferator-activated receptor gamma[83]
- PIK3R3[96]
- Plasminogen activator inhibitor-2[97]
- Polymerase (DNA directed), alpha 1[98]
- PRDM2[99]
- PRKRA[100]
- Prohibitin[58][101]
- Promyelocytic leukemia protein[102]
- RBBP4[65][103]
- RBBP7[53][103]
- RBBP8[77][104]
- RBBP9[105]
- SNAPC1[106]
- SKP2[107][108]
- SNAPC3[106]
- SNW1[109]
- SUV39H1[110][111]
- TAF1[62][112][113][114]
- THOC1[115]
- TRAP1[116]
- TRIP11[117]
- UBTF[118]
- USP4.[119]
Detection
Several methods for detecting the RB1 gene mutations have been developed[120] including a method that can detect large deletions that correlate with advanced stage retinoblastoma.[121]
See also
- p53 - involved in the DNA repair support function of pRb
- Transcription coregulator
- Retinoblastoma
References
- GRCh38: Ensembl release 89: ENSG00000139687 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000022105 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Murphree AL, Benedict WF (March 1984). "Retinoblastoma: clues to human oncogenesis". Science. 223 (4640): 1028–33. Bibcode:1984Sci...223.1028L. doi:10.1126/science.6320372. PMID 6320372.
- Shao Z, Robbins PD (January 1995). "Differential regulation of E2F and Sp1-mediated transcription by G1 cyclins". Oncogene. 10 (2): 221–8. PMID 7838522.
- Korenjak M, Brehm A (October 2005). "E2F-Rb complexes regulating transcription of genes important for differentiation and development". Current Opinion in Genetics & Development. 15 (5): 520–7. doi:10.1016/j.gde.2005.07.001. PMID 16081278.
- Münger K, Howley PM (November 2002). "Human papillomavirus immortalization and transformation functions". Virus Research. 89 (2): 213–28. doi:10.1016/S0168-1702(02)00190-9. PMID 12445661.
- Gallego J (May 2016). "Multicellular Life Was Caused By The Same Gene That Suppresses Cancer". Kansas State University.
- Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, et al. (April 2005). "Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up". Journal of Clinical Oncology. 23 (10): 2272–9. doi:10.1200/JCO.2005.05.054. PMID 15800318.
- Knudson AG (April 1971). "Mutation and cancer: statistical study of retinoblastoma". Proceedings of the National Academy of Sciences of the United States of America. 68 (4): 820–3. Bibcode:1971PNAS...68..820K. doi:10.1073/pnas.68.4.820. PMC 389051. PMID 5279523.
- "OrthoMaM phylogenetic marker: RB1 coding sequence". Archived from the original on 2015-09-24. Retrieved 2009-12-02.
- Frolov MV, Dyson NJ (May 2004). "Molecular mechanisms of E2F-dependent activation and pRB-mediated repression". Journal of Cell Science. 117 (Pt 11): 2173–81. doi:10.1242/jcs.01227. PMID 15126619.
- Morris EJ, Dyson NJ (2001). "Retinoblastoma protein partners". Advances in Cancer Research. 82: 1–54. doi:10.1016/S0065-230X(01)82001-7. ISBN 9780120066827. PMID 11447760.
- Dick FA, Rubin SM (May 2013). "Molecular mechanisms underlying pRB protein function". Nature Reviews. Molecular Cell Biology. 14 (5): 297–306. doi:10.1038/nrm3567. PMC 4754300. PMID 23594950.
- Goodrich DW, Wang NP, Qian YW, Lee EY, Lee WH (October 1991). "The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle". Cell. 67 (2): 293–302. doi:10.1016/0092-8674(91)90181-w. PMID 1655277. S2CID 12990398.
- Wu CL, Zukerberg LR, Ngwu C, Harlow E, Lees JA (May 1995). "In vivo association of E2F and DP family proteins". Molecular and Cellular Biology. 15 (5): 2536–46. doi:10.1128/mcb.15.5.2536. PMC 230484. PMID 7739537.
- Funk JO, Waga S, Harry JB, Espling E, Stillman B, Galloway DA (August 1997). "Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein". Genes & Development. 11 (16): 2090–100. doi:10.1016/0168-9525(97)90029-9. PMC 316456. PMID 9284048.
- De Veylder L, Joubès J, Inzé D (December 2003). "Plant cell cycle transitions". Current Opinion in Plant Biology. 6 (6): 536–43. doi:10.1016/j.pbi.2003.09.001. PMID 14611951.
- de Jager SM, Maughan S, Dewitte W, Scofield S, Murray JA (June 2005). "The developmental context of cell-cycle control in plants". Seminars in Cell & Developmental Biology. 16 (3): 385–96. doi:10.1016/j.semcdb.2005.02.004. PMID 15840447.
- Greenblatt RJ (2005). "Human papillomaviruses: Diseases, diagnosis, and a possible vaccine". Clinical Microbiology Newsletter. 27 (18): 139–45. doi:10.1016/j.clinmicnews.2005.09.001.
- Sinal SH, Woods CR (October 2005). "Human papillomavirus infections of the genital and respiratory tracts in young children". Seminars in Pediatric Infectious Diseases. 16 (4): 306–16. doi:10.1053/j.spid.2005.06.010. PMID 16210110.
- Das SK, Hashimoto T, Shimizu K, Yoshida T, Sakai T, Sowa Y, et al. (November 2005). "Fucoxanthin induces cell cycle arrest at G0/G1 phase in human colon carcinoma cells through up-regulation of p21WAF1/Cip1". Biochimica et Biophysica Acta (BBA) - General Subjects. 1726 (3): 328–35. doi:10.1016/j.bbagen.2005.09.007. PMID 16236452.
- Bartkova J, Grøn B, Dabelsteen E, Bartek J (February 2003). "Cell-cycle regulatory proteins in human wound healing". Archives of Oral Biology. 48 (2): 125–32. doi:10.1016/S0003-9969(02)00202-9. PMID 12642231.
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF (June 2014). "Cyclin D activates the Rb tumor suppressor by mono-phosphorylation". eLife. 3. doi:10.7554/eLife.02872. PMC 4076869. PMID 24876129.
- Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, et al. (March 2019). "A Code of Mono-phosphorylation Modulates the Function of RB". Molecular Cell. 73 (5): 985–1000.e6. doi:10.1016/j.molcel.2019.01.004. PMC 6424368. PMID 30711375.
- Vietri M, Bianchi M, Ludlow JW, Mittnacht S, Villa-Moruzzi E (February 2006). "Direct interaction between the catalytic subunit of Protein Phosphatase 1 and pRb". Cancer Cell International. 6: 3. doi:10.1186/1475-2867-6-3. PMC 1382259. PMID 16466572.
- Burkhart DL, Sage J (September 2008). "Cellular mechanisms of tumour suppression by the retinoblastoma gene". Nature Reviews. Cancer. 8 (9): 671–82. doi:10.1038/nrc2399. PMC 6996492. PMID 18650841.
- Seyfried TN, Shelton LM (2010). "Cancer as a metabolic disease". Nutrition & Metabolism. 7: 7. doi:10.1186/1743-7075-7-7. PMC 2845135. PMID 20181022.
- Zacksenhaus E, Shrestha M, Liu JC, Jiang Z (2017). "Mitochondrial OXPHOS Induced by RB1 Deficiency in Breast Cancer: Implications for Anabolic Metabolism, Stemness, and Metastasis". Trends in Cancer. 3 (11): 768–779. doi:10.1016/j.trecan.2017.09.002. PMID 29120753.
- Sage J (July 2012). "The retinoblastoma tumor suppressor and stem cell biology". Genes & Development. 26 (13): 1409–20. doi:10.1101/gad.193730.112. PMC 3403009. PMID 22751497.
- Dick FA, Goodrich DW, Sage J, Dyson NJ (July 2018). "Non-canonical functions of the RB protein in cancer". Nature Reviews. Cancer. 18 (7): 442–451. doi:10.1038/s41568-018-0008-5. PMC 6693677. PMID 29692417.
- Dyson NJ (July 2016). "RB1: a prototype tumor suppressor and an enigma". Genes & Development. 30 (13): 1492–502. doi:10.1101/gad.282145.116. PMC 4949322. PMID 27401552.
- Knudsen ES, Wang JY (February 2010). "Targeting the RB-pathway in cancer therapy". Clinical Cancer Research. 16 (4): 1094–9. doi:10.1158/1078-0432.CCR-09-0787. PMC 2822892. PMID 20145169.
- Bui TB, Burgers DM, Agterof MJ, van de Garde EM (2019). "Real-World Effectiveness of Palbociclib Versus Clinical Trial Results in Patients With Advanced/Metastatic Breast Cancer That Progressed on Previous Endocrine Therapy". Breast Cancer. 13: 1178223418823238. doi:10.1177/1178223418823238. PMC 6330732. PMID 30675102.
- Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, et al. (July 2016). "Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors". Cancer Discovery. 6 (7): 740–53. doi:10.1158/2159-8290.CD-16-0095. PMID 27217383.
- Walter DM, Yates TJ, Ruiz-Torres M, Kim-Kiselak C, Gudiel AA, Deshpande C, et al. (May 2019). "RB constrains lineage fidelity and multiple stages of tumour progression and metastasis". Nature. 569 (7756): 423–427. Bibcode:2019Natur.569..423W. doi:10.1038/s41586-019-1172-9. PMC 6522292. PMID 31043741.
- Pomerantz JH, Blau HM (June 2013). "Tumor suppressors: enhancers or suppressors of regeneration?". Development. 140 (12): 2502–12. doi:10.1242/dev.084210. PMC 3666379. PMID 23715544.
- Sage C, Huang M, Vollrath MA, Brown MC, Hinds PW, Corey DP, et al. (May 2006). "Essential role of retinoblastoma protein in mammalian hair cell development and hearing". Proceedings of the National Academy of Sciences of the United States of America. 103 (19): 7345–50. Bibcode:2006PNAS..103.7345S. doi:10.1073/pnas.0510631103. PMC 1450112. PMID 16648263.
- Weber T, Corbett MK, Chow LM, Valentine MB, Baker SJ, Zuo J (January 2008). "Rapid cell-cycle reentry and cell death after acute inactivation of the retinoblastoma gene product in postnatal cochlear hair cells". Proceedings of the National Academy of Sciences of the United States of America. 105 (2): 781–5. Bibcode:2008PNAS..105..781W. doi:10.1073/pnas.0708061105. PMC 2206613. PMID 18178626.
- Lu N, Chen Y, Wang Z, Chen G, Lin Q, Chen ZY, Li H (January 2013). "Sonic hedgehog initiates cochlear hair cell regeneration through downregulation of retinoblastoma protein". Biochemical and Biophysical Research Communications. 430 (2): 700–5. doi:10.1016/j.bbrc.2012.11.088. PMC 3579567. PMID 23211596.
- Christie KJ, Krishnan A, Martinez JA, Purdy K, Singh B, Eaton S, Zochodne D (April 2014). "Enhancing adult nerve regeneration through the knockdown of retinoblastoma protein". Nature Communications. 5: 3670. Bibcode:2014NatCo...5.3670C. doi:10.1038/ncomms4670. PMC 5028199. PMID 24752312.
- Miyamura T, Nishimura J, Yufu Y, Nawata H (February 1997). "Interaction of BCR-ABL with the retinoblastoma protein in Philadelphia chromosome-positive cell lines". International Journal of Hematology. 65 (2): 115–21. doi:10.1016/S0925-5710(96)00539-7. PMID 9071815.
- Welch PJ, Wang JY (November 1993). "A C-terminal protein-binding domain in the retinoblastoma protein regulates nuclear c-Abl tyrosine kinase in the cell cycle". Cell. 75 (4): 779–90. doi:10.1016/0092-8674(93)90497-E. PMID 8242749.
- Lu J, Danielsen M (November 1998). "Differential regulation of androgen and glucocorticoid receptors by retinoblastoma protein". The Journal of Biological Chemistry. 273 (47): 31528–33. doi:10.1074/jbc.273.47.31528. PMID 9813067.
- Yeh S, Miyamoto H, Nishimura K, Kang H, Ludlow J, Hsiao P, et al. (July 1998). "Retinoblastoma, a tumor suppressor, is a coactivator for the androgen receptor in human prostate cancer DU145 cells". Biochemical and Biophysical Research Communications. 248 (2): 361–7. doi:10.1006/bbrc.1998.8974. PMID 9675141.
- Bruno T, De Angelis R, De Nicola F, Barbato C, Di Padova M, Corbi N, et al. (November 2002). "Che-1 affects cell growth by interfering with the recruitment of HDAC1 by Rb". Cancer Cell. 2 (5): 387–99. doi:10.1016/S1535-6108(02)00182-4. PMID 12450794.
- Fanciulli M, Bruno T, Di Padova M, De Angelis R, Iezzi S, Iacobini C, et al. (May 2000). "Identification of a novel partner of RNA polymerase II subunit 11, Che-1, which interacts with and affects the growth suppression function of Rb". FASEB Journal. 14 (7): 904–12. doi:10.1096/fasebj.14.7.904. PMID 10783144. S2CID 43175069.
- Lai A, Lee JM, Yang WM, DeCaprio JA, Kaelin WG, Seto E, Branton PE (October 1999). "RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins". Molecular and Cellular Biology. 19 (10): 6632–41. doi:10.1128/mcb.19.10.6632. PMC 84642. PMID 10490602.
- Ge NL, Elferink CJ (August 1998). "A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle". The Journal of Biological Chemistry. 273 (35): 22708–13. doi:10.1074/jbc.273.35.22708. PMID 9712901.
- Aprelikova ON, Fang BS, Meissner EG, Cotter S, Campbell M, Kuthiala A, et al. (October 1999). "BRCA1-associated growth arrest is RB-dependent". Proceedings of the National Academy of Sciences of the United States of America. 96 (21): 11866–71. Bibcode:1999PNAS...9611866A. doi:10.1073/pnas.96.21.11866. PMC 18378. PMID 10518542.
- Fan S, Yuan R, Ma YX, Xiong J, Meng Q, Erdos M, et al. (August 2001). "Disruption of BRCA1 LXCXE motif alters BRCA1 functional activity and regulation of RB family but not RB protein binding". Oncogene. 20 (35): 4827–41. doi:10.1038/sj.onc.1204666. PMID 11521194.
- Yarden RI, Brody LC (April 1999). "BRCA1 interacts with components of the histone deacetylase complex". Proceedings of the National Academy of Sciences of the United States of America. 96 (9): 4983–8. Bibcode:1999PNAS...96.4983Y. doi:10.1073/pnas.96.9.4983. PMC 21803. PMID 10220405.
- Johnston IM, Allison SJ, Morton JP, Schramm L, Scott PH, White RJ (June 2002). "CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells". Molecular and Cellular Biology. 22 (11): 3757–68. doi:10.1128/MCB.22.11.3757-3768.2002. PMC 133823. PMID 11997511.
- Sutcliffe JE, Cairns CA, McLees A, Allison SJ, Tosh K, White RJ (June 1999). "RNA polymerase III transcription factor IIIB is a target for repression by pocket proteins p107 and p130". Molecular and Cellular Biology. 19 (6): 4255–61. doi:10.1128/mcb.19.6.4255. PMC 104385. PMID 10330166.
- Nishitani J, Nishinaka T, Cheng CH, Rong W, Yokoyama KK, Chiu R (February 1999). "Recruitment of the retinoblastoma protein to c-Jun enhances transcription activity mediated through the AP-1 binding site". The Journal of Biological Chemistry. 274 (9): 5454–61. doi:10.1074/jbc.274.9.5454. PMID 10026157.
- Wang S, Ghosh RN, Chellappan SP (December 1998). "Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation". Molecular and Cellular Biology. 18 (12): 7487–98. doi:10.1128/mcb.18.12.7487. PMC 109329. PMID 9819434.
- Wang S, Nath N, Fusaro G, Chellappan S (November 1999). "Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals". Molecular and Cellular Biology. 19 (11): 7447–60. doi:10.1128/mcb.19.11.7447. PMC 84738. PMID 10523633.
- Simone C, Bagella L, Bellan C, Giordano A (June 2002). "Physical interaction between pRb and cdk9/cyclinT2 complex". Oncogene. 21 (26): 4158–65. doi:10.1038/sj.onc.1205511. PMID 12037672.
- Gupta S, Luong MX, Bleuming SA, Miele A, Luong M, Young D, et al. (September 2003). "Tumor suppressor pRB functions as a co-repressor of the CCAAT displacement protein (CDP/cut) to regulate cell cycle controlled histone H4 transcription". Journal of Cellular Physiology. 196 (3): 541–56. doi:10.1002/jcp.10335. PMID 12891711. S2CID 2287673.
- Yang R, Müller C, Huynh V, Fung YK, Yee AS, Koeffler HP (March 1999). "Functions of cyclin A1 in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins". Molecular and Cellular Biology. 19 (3): 2400–7. doi:10.1128/mcb.19.3.2400. PMC 84032. PMID 10022926.
- Siegert JL, Rushton JJ, Sellers WR, Kaelin WG, Robbins PD (November 2000). "Cyclin D1 suppresses retinoblastoma protein-mediated inhibition of TAFII250 kinase activity". Oncogene. 19 (50): 5703–11. doi:10.1038/sj.onc.1203966. PMID 11126356.
- Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA (May 1993). "Physical interaction of the retinoblastoma protein with human D cyclins". Cell. 73 (3): 499–511. doi:10.1016/0092-8674(93)90137-F. PMID 8490963. S2CID 24708871.
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP (July 2000). "DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters". Nature Genetics. 25 (3): 338–42. doi:10.1038/77124. PMID 10888886. S2CID 10983932.
- Nicolas E, Ait-Si-Ali S, Trouche D (August 2001). "The histone deacetylase HDAC3 targets RbAp48 to the retinoblastoma protein". Nucleic Acids Research. 29 (15): 3131–6. doi:10.1093/nar/29.15.3131. PMC 55834. PMID 11470869.
- Pardo PS, Leung JK, Lucchesi JC, Pereira-Smith OM (December 2002). "MRG15, a novel chromodomain protein, is present in two distinct multiprotein complexes involved in transcriptional activation". The Journal of Biological Chemistry. 277 (52): 50860–6. doi:10.1074/jbc.M203839200. PMID 12397079.
- Choubey D, Li SJ, Datta B, Gutterman JU, Lengyel P (October 1996). "Inhibition of E2F-mediated transcription by p202". The EMBO Journal. 15 (20): 5668–78. doi:10.1002/j.1460-2075.1996.tb00951.x. PMC 452311. PMID 8896460.
- Fajas L, Paul C, Zugasti O, Le Cam L, Polanowska J, Fabbrizio E, et al. (July 2000). "pRB binds to and modulates the transrepressing activity of the E1A-regulated transcription factor p120E4F". Proceedings of the National Academy of Sciences of the United States of America. 97 (14): 7738–43. Bibcode:2000PNAS...97.7738F. doi:10.1073/pnas.130198397. PMC 16614. PMID 10869426.
- Dyson N, Dembski M, Fattaey A, Ngwu C, Ewen M, Helin K (December 1993). "Analysis of p107-associated proteins: p107 associates with a form of E2F that differs from pRB-associated E2F-1". Journal of Virology. 67 (12): 7641–7. doi:10.1128/JVI.67.12.7641-7647.1993. PMC 238233. PMID 8230483.
- Taniura H, Taniguchi N, Hara M, Yoshikawa K (January 1998). "Necdin, a postmitotic neuron-specific growth suppressor, interacts with viral transforming proteins and cellular transcription factor E2F1". The Journal of Biological Chemistry. 273 (2): 720–8. doi:10.1074/jbc.273.2.720. PMID 9422723.
- Lee C, Chang JH, Lee HS, Cho Y (December 2002). "Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor". Genes & Development. 16 (24): 3199–212. doi:10.1101/gad.1046102. PMC 187509. PMID 12502741.
- Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, et al. (December 2000). "Cells degrade a novel inhibitor of differentiation with E1A-like properties upon exiting the cell cycle". Molecular and Cellular Biology. 20 (23): 8889–902. doi:10.1128/MCB.20.23.8889-8902.2000. PMC 86544. PMID 11073989.
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD (December 2000). "A novel Rb- and p300-binding protein inhibits transactivation by MyoD". Molecular and Cellular Biology. 20 (23): 8903–15. doi:10.1128/MCB.20.23.8903-8915.2000. PMC 86545. PMID 11073990.
- Kim TA, Lim J, Ota S, Raja S, Rogers R, Rivnay B, et al. (May 1998). "NRP/B, a novel nuclear matrix protein, associates with p110(RB) and is involved in neuronal differentiation". The Journal of Cell Biology. 141 (3): 553–66. doi:10.1083/jcb.141.3.553. PMC 2132755. PMID 9566959.
- Craven RJ, Cance WG, Liu ET (September 1995). "The nuclear tyrosine kinase Rak associates with the retinoblastoma protein pRb". Cancer Research. 55 (18): 3969–72. PMID 7664264.
- Lavender P, Vandel L, Bannister AJ, Kouzarides T (June 1997). "The HMG-box transcription factor HBP1 is targeted by the pocket proteins and E1A". Oncogene. 14 (22): 2721–8. doi:10.1038/sj.onc.1201243. PMID 9178770.
- Dick FA, Sailhamer E, Dyson NJ (May 2000). "Mutagenesis of the pRB pocket reveals that cell cycle arrest functions are separable from binding to viral oncoproteins". Molecular and Cellular Biology. 20 (10): 3715–27. doi:10.1128/MCB.20.10.3715-3727.2000. PMC 85672. PMID 10779361.
- Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T (January 2000). "DNA methyltransferase Dnmt1 associates with histone deacetylase activity". Nature Genetics. 24 (1): 88–91. doi:10.1038/71750. PMID 10615135. S2CID 20428600.
- Puri PL, Iezzi S, Stiegler P, Chen TT, Schiltz RL, Muscat GE, et al. (October 2001). "Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis". Molecular Cell. 8 (4): 885–97. doi:10.1016/S1097-2765(01)00373-2. PMID 11684023.
- Wang S, Fusaro G, Padmanabhan J, Chellappan SP (December 2002). "Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression". Oncogene. 21 (55): 8388–96. doi:10.1038/sj.onc.1205944. PMID 12466959.
- Luo RX, Postigo AA, Dean DC (February 1998). "Rb interacts with histone deacetylase to repress transcription". Cell. 92 (4): 463–73. doi:10.1016/S0092-8674(00)80940-X. PMID 9491888. S2CID 18857544.
- Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A, Trouche D (September 1998). "The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase". Proceedings of the National Academy of Sciences of the United States of America. 95 (18): 10493–8. Bibcode:1998PNAS...9510493F. doi:10.1073/pnas.95.18.10493. PMC 27922. PMID 9724731.
- Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, et al. (December 2002). "The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation". Developmental Cell. 3 (6): 903–10. doi:10.1016/S1534-5807(02)00360-X. PMID 12479814.
- Radulescu RT, Bellitti MR, Ruvo M, Cassani G, Fassina G (January 1995). "Binding of the LXCXE insulin motif to a hexapeptide derived from retinoblastoma protein". Biochemical and Biophysical Research Communications. 206 (1): 97–102. doi:10.1006/bbrc.1995.1014. PMID 7818556.
- Chan SW, Hong W (July 2001). "Retinoblastoma-binding protein 2 (Rbp2) potentiates nuclear hormone receptor-mediated transcription". The Journal of Biological Chemistry. 276 (30): 28402–12. doi:10.1074/jbc.M100313200. PMID 11358960.
- Kim YW, Otterson GA, Kratzke RA, Coxon AB, Kaye FJ (November 1994). "Differential specificity for binding of retinoblastoma binding protein 2 to RB, p107, and TATA-binding protein". Molecular and Cellular Biology. 14 (11): 7256–64. doi:10.1128/mcb.14.11.7256. PMC 359260. PMID 7935440.
- An P, Sáenz Robles MT, Pipas JM (13 October 2012). "Large T antigens of polyomaviruses: amazing molecular machines". Annual Review of Microbiology. 66 (1): 213–236. doi:10.1146/annurev-micro-092611-150154. PMID 22994493.
- Arora R, Chang Y, Moore PS (August 2012). "MCV and Merkel cell carcinoma: a molecular success story". Current Opinion in Virology. 2 (4): 489–498. doi:10.1016/j.coviro.2012.05.007. PMC 3422445. PMID 22710026.
- Gagrica S, Hauser S, Kolfschoten I, Osterloh L, Agami R, Gaubatz S (November 2004). "Inhibition of oncogenic transformation by mammalian Lin-9, a pRB-associated protein". The EMBO Journal. 23 (23): 4627–38. doi:10.1038/sj.emboj.7600470. PMC 533054. PMID 15538385.
- Sterner JM, Dew-Knight S, Musahl C, Kornbluth S, Horowitz JM (May 1998). "Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7". Molecular and Cellular Biology. 18 (5): 2748–57. doi:10.1128/mcb.18.5.2748. PMC 110654. PMID 9566894.
- Leung JK, Berube N, Venable S, Ahmed S, Timchenko N, Pereira-Smith OM (October 2001). "MRG15 activates the B-myb promoter through formation of a nuclear complex with the retinoblastoma protein and the novel protein PAM14". The Journal of Biological Chemistry. 276 (42): 39171–8. doi:10.1074/jbc.M103435200. PMID 11500496.
- Mal A, Sturniolo M, Schiltz RL, Ghosh MK, Harter ML (April 2001). "A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program". The EMBO Journal. 20 (7): 1739–53. doi:10.1093/emboj/20.7.1739. PMC 145490. PMID 11285237.
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B (February 1993). "Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation". Cell. 72 (3): 309–24. doi:10.1016/0092-8674(93)90110-C. PMID 8381715. S2CID 21581966.
- Goo YH, Na SY, Zhang H, Xu J, Hong S, Cheong J, et al. (February 2004). "Interactions between activating signal cointegrator-2 and the tumor suppressor retinoblastoma in androgen receptor transactivation". The Journal of Biological Chemistry. 279 (8): 7131–5. doi:10.1074/jbc.M312563200. PMID 14645241.
- Xia X, Cheng A, Lessor T, Zhang Y, Hamburger AW (May 2001). "Ebp1, an ErbB-3 binding protein, interacts with Rb and affects Rb transcriptional regulation". Journal of Cellular Physiology. 187 (2): 209–17. doi:10.1002/jcp.1075. PMID 11268000. S2CID 42721280.
- Xia X, Cheng A, Akinmade D, Hamburger AW (March 2003). "The N-terminal 24 amino acids of the p55 gamma regulatory subunit of phosphoinositide 3-kinase binds Rb and induces cell cycle arrest". Molecular and Cellular Biology. 23 (5): 1717–25. doi:10.1128/MCB.23.5.1717-1725.2003. PMC 151709. PMID 12588990.
- Darnell GA, Antalis TM, Johnstone RW, Stringer BW, Ogbourne SM, Harrich D, Suhrbier A (September 2003). "Inhibition of retinoblastoma protein degradation by interaction with the serpin plasminogen activator inhibitor 2 via a novel consensus motif". Molecular and Cellular Biology. 23 (18): 6520–32. doi:10.1128/MCB.23.18.6520-6532.2003. PMC 193706. PMID 12944478.
- Takemura M, Kitagawa T, Izuta S, Wasa J, Takai A, Akiyama T, Yoshida S (November 1997). "Phosphorylated retinoblastoma protein stimulates DNA polymerase alpha". Oncogene. 15 (20): 2483–92. doi:10.1038/sj.onc.1201431. PMID 9395244.
- Buyse IM, Shao G, Huang S (May 1995). "The retinoblastoma protein binds to RIZ, a zinc-finger protein that shares an epitope with the adenovirus E1A protein". Proceedings of the National Academy of Sciences of the United States of America. 92 (10): 4467–71. Bibcode:1995PNAS...92.4467B. doi:10.1073/pnas.92.10.4467. PMC 41965. PMID 7538672.
- Simons A, Melamed-Bessudo C, Wolkowicz R, Sperling J, Sperling R, Eisenbach L, Rotter V (January 1997). "PACT: cloning and characterization of a cellular p53 binding protein that interacts with Rb". Oncogene. 14 (2): 145–55. doi:10.1038/sj.onc.1200825. PMID 9010216.
- Wang S, Nath N, Adlam M, Chellappan S (June 1999). "Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function". Oncogene. 18 (23): 3501–10. doi:10.1038/sj.onc.1202684. PMID 10376528.
- Alcalay M, Tomassoni L, Colombo E, Stoldt S, Grignani F, Fagioli M, et al. (February 1998). "The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein". Molecular and Cellular Biology. 18 (2): 1084–93. doi:10.1128/mcb.18.2.1084. PMC 108821. PMID 9448006.
- Qian YW, Lee EY (October 1995). "Dual retinoblastoma-binding proteins with properties related to a negative regulator of ras in yeast". The Journal of Biological Chemistry. 270 (43): 25507–13. doi:10.1074/jbc.270.43.25507. PMID 7503932.
- Fusco C, Reymond A, Zervos AS (August 1998). "Molecular cloning and characterization of a novel retinoblastoma-binding protein". Genomics. 51 (3): 351–8. doi:10.1006/geno.1998.5368. PMID 9721205.
- Woitach JT, Zhang M, Niu CH, Thorgeirsson SS (August 1998). "A retinoblastoma-binding protein that affects cell-cycle control and confers transforming ability". Nature Genetics. 19 (4): 371–4. doi:10.1038/1258. PMID 9697699. S2CID 11374970.
- Hirsch HA, Gu L, Henry RW (December 2000). "The retinoblastoma tumor suppressor protein targets distinct general transcription factors to regulate RNA polymerase III gene expression". Molecular and Cellular Biology. 20 (24): 9182–91. doi:10.1128/MCB.20.24.9182-9191.2000. PMC 102176. PMID 11094070.
- Ji P, Jiang H, Rekhtman K, Bloom J, Ichetovkin M, Pagano M, Zhu L (October 2004). "An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant". Molecular Cell. 16 (1): 47–58. doi:10.1016/j.molcel.2004.09.029. PMID 15469821.
- Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, et al. (January 2010). "Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice". Nature Genetics. 42 (1): 83–8. doi:10.1038/ng.498. PMC 2990528. PMID 19966802.
- Prathapam T, Kühne C, Banks L (December 2002). "Skip interacts with the retinoblastoma tumor suppressor and inhibits its transcriptional repression activity". Nucleic Acids Research. 30 (23): 5261–8. doi:10.1093/nar/gkf658. PMC 137971. PMID 12466551.
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, et al. (August 2001). "Rb targets histone H3 methylation and HP1 to promoters". Nature. 412 (6846): 561–5. Bibcode:2001Natur.412..561N. doi:10.1038/35087620. PMID 11484059. S2CID 4378296.
- Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D (October 2001). "Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase". Molecular and Cellular Biology. 21 (19): 6484–94. doi:10.1128/MCB.21.19.6484-6494.2001. PMC 99795. PMID 11533237.
- Shao Z, Ruppert S, Robbins PD (April 1995). "The retinoblastoma-susceptibility gene product binds directly to the human TATA-binding protein-associated factor TAFII250". Proceedings of the National Academy of Sciences of the United States of America. 92 (8): 3115–9. Bibcode:1995PNAS...92.3115S. doi:10.1073/pnas.92.8.3115. PMC 42115. PMID 7724524.
- Siegert JL, Robbins PD (January 1999). "Rb inhibits the intrinsic kinase activity of TATA-binding protein-associated factor TAFII250". Molecular and Cellular Biology. 19 (1): 846–54. doi:10.1128/MCB.19.1.846. PMC 83941. PMID 9858607.
- Shao Z, Siegert JL, Ruppert S, Robbins PD (July 1997). "Rb interacts with TAF(II)250/TFIID through multiple domains". Oncogene. 15 (4): 385–92. doi:10.1038/sj.onc.1201204. PMID 9242374.
- Durfee T, Mancini MA, Jones D, Elledge SJ, Lee WH (November 1994). "The amino-terminal region of the retinoblastoma gene product binds a novel nuclear matrix protein that co-localizes to centers for RNA processing". The Journal of Cell Biology. 127 (3): 609–22. doi:10.1083/jcb.127.3.609. PMC 2120229. PMID 7525595.
- Chen CF, Chen Y, Dai K, Chen PL, Riley DJ, Lee WH (September 1996). "A new member of the hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock". Molecular and Cellular Biology. 16 (9): 4691–9. doi:10.1128/MCB.16.9.4691. PMC 231469. PMID 8756626.
- Chang KH, Chen Y, Chen TT, Chou WH, Chen PL, Ma YY, et al. (August 1997). "A thyroid hormone receptor coactivator negatively regulated by the retinoblastoma protein". Proceedings of the National Academy of Sciences of the United States of America. 94 (17): 9040–5. Bibcode:1997PNAS...94.9040C. doi:10.1073/pnas.94.17.9040. PMC 23019. PMID 9256431.
- Hannan KM, Hannan RD, Smith SD, Jefferson LS, Lun M, Rothblum LI (October 2000). "Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1". Oncogene. 19 (43): 4988–99. doi:10.1038/sj.onc.1203875. PMID 11042686.
- Blanchette P, Gilchrist CA, Baker RT, Gray DA (September 2001). "Association of UNP, a ubiquitin-specific protease, with the pocket proteins pRb, p107 and p130". Oncogene. 20 (39): 5533–7. doi:10.1038/sj.onc.1204823. PMID 11571651.
- Parsam VL, Kannabiran C, Honavar S, Vemuganti GK, Ali MJ (December 2009). "A comprehensive, sensitive and economical approach for the detection of mutations in the RB1 gene in retinoblastoma" (PDF). Journal of Genetics. 88 (4): 517–27. doi:10.1007/s12041-009-0069-z. PMID 20090211. S2CID 10723496.
- Ali MJ, Parsam VL, Honavar SG, Kannabiran C, Vemuganti GK, Reddy VA (October 2010). "RB1 gene mutations in retinoblastoma and its clinical correlation". Saudi Journal of Ophthalmology. 24 (4): 119–23. doi:10.1016/j.sjopt.2010.05.003. PMC 3729507. PMID 23960888.
Further reading
- Momand J, Wu HH, Dasgupta G (January 2000). "MDM2--master regulator of the p53 tumor suppressor protein". Gene. 242 (1–2): 15–29. doi:10.1016/S0378-1119(99)00487-4. PMID 10721693.
- Zheng L, Lee WH (2003). "Retinoblastoma tumor suppressor and genome stability". Advances in Cancer Research Volume 85. Advances in Cancer Research. Vol. 85. pp. 13–50. doi:10.1016/S0065-230X(02)85002-3. ISBN 978-0-12-006685-8. PMID 12374284.
- Classon M, Harlow E (December 2002). "The retinoblastoma tumour suppressor in development and cancer". Nature Reviews. Cancer. 2 (12): 910–7. doi:10.1038/nrc950. PMID 12459729. S2CID 22937378.
- Lai H, Ma F, Lai S (January 2003). "Identification of the novel role of pRB in eye cancer". Journal of Cellular Biochemistry. 88 (1): 121–7. doi:10.1002/jcb.10283. PMID 12461781. S2CID 34538683.
- Simin K, Wu H, Lu L, Pinkel D, Albertson D, Cardiff RD, Van Dyke T (February 2004). "pRb inactivation in mammary cells reveals common mechanisms for tumor initiation and progression in divergent epithelia". PLOS Biology. 2 (2): E22. doi:10.1371/journal.pbio.0020022. PMC 340938. PMID 14966529.
- Lohmann DR, Gallie BL (August 2004). "Retinoblastoma: revisiting the model prototype of inherited cancer". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 129C (1): 23–8. doi:10.1002/ajmg.c.30024. PMID 15264269. S2CID 41922148.
- Clemo NK, Arhel NJ, Barnes JD, Baker J, Moorghen M, Packham GK, et al. (August 2005). "The role of the retinoblastoma protein (Rb) in the nuclear localization of BAG-1: implications for colorectal tumour cell survival". Biochemical Society Transactions. 33 (Pt 4): 676–8. doi:10.1042/BST0330676. PMID 16042572.
- Rodríguez-Cruz M, del Prado M, Salcedo M (2006). "[Genomic retinoblastoma perspectives: implications of tumor supressor [sic] gene RB1]". Revista de Investigacion Clinica. 57 (4): 572–81. PMID 16315642.
- Knudsen ES, Knudsen KE (July 2006). "Retinoblastoma tumor suppressor: where cancer meets the cell cycle". Experimental Biology and Medicine. 231 (7): 1271–81. doi:10.1177/153537020623100713. PMID 16816134. S2CID 29078799.
External links
- RB1+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- Retinoblastoma+genes at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- GeneReviews/NIH/NCBI/UW entry on Retinoblastoma
- Retinoblastoma Genetics
- Drosophila Retinoblastoma-family protein - The Interactive Fly
- Drosophila Retinoblastoma-family protein 2 - The Interactive Fly
- Evolutionary Homologs Retinoblastoma-family proteins - The Interactive Fly
- There is a diagram of the pRb-E2F interactions here.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|



{kind=link}