Sodium channel
Sodium channels are integral membrane proteins that form ion channels, conducting sodium ions (Na+) through a cell's membrane.[1][2] They belong to the superfamily of cation channels and can be classified according to the trigger that opens the channel for such ions, i.e. either a voltage-change ("voltage-gated", "voltage-sensitive", or "voltage-dependent" sodium channel; also called "VGSCs" or "Nav channel") or a binding of a substance (a ligand) to the channel (ligand-gated sodium channels).
In excitable cells such as neurons, myocytes, and certain types of glia, sodium channels are responsible for the rising phase of action potentials. These channels go through three different states called resting, active and inactive states. Even though the resting and inactive states would not allow the ions to flow through the channels the difference exists with respect to their structural conformation.
Selectivity
Sodium channels are highly selective for the transport of ions across cell membranes. The high selectivity with respect to the sodium ion is achieved in many different ways. All involve encapsulation of the sodium ion in a cavity of specific size within a larger molecule.[3]
Voltage-gated sodium channels
Structure
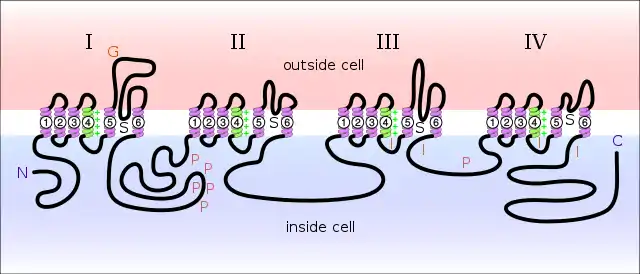
Sodium channels consist of large alpha subunits that associate with proteins, such as beta subunits. An alpha subunit forms the core of the channel and is functional on its own. When the alpha subunit protein is expressed by a cell, it is able to form channels that conduct Na+ in a voltage-gated way, even if beta subunits or other known modulating proteins are not expressed. When accessory proteins assemble with α subunits, the resulting complex can display altered voltage dependence and cellular localization.
The alpha subunit has four repeat domains, labelled I through IV, each containing six membrane-spanning segments, labelled S1 through S6. The highly conserved S4 segment acts as the channel's voltage sensor. The voltage sensitivity of this channel is due to positive amino acids located at every third position.[5] When stimulated by a change in transmembrane voltage, this segment moves toward the extracellular side of the cell membrane, allowing the channel to become permeable to ions. The ions are conducted through a pore, which can be broken into two regions. The more external (i.e., more extracellular) portion of the pore is formed by the "P-loops" (the region between S5 and S6) of the four domains. This region is the most narrow part of the pore and is responsible for its ion selectivity. The inner portion (i.e., more cytoplasmic) of the pore is formed by the combined S5 and S6 segments of the four domains. The region linking domains III and IV is also important for channel function. This region plugs the channel after prolonged activation, inactivating it.
Gating
Voltage-gated Na+ channels have three main conformational states: closed, open and inactivated. Forward/back transitions between these states are correspondingly referred to as activation/deactivation (between open and closed, respectively), inactivation/reactivation (between inactivated and open, respectively), and recovery from inactivation/closed-state inactivation (between inactivated and closed, respectively). Closed and inactivated states are ion impermeable.
Before an action potential occurs, the axonal membrane is at its normal resting potential, about −70 mV in most human neurons, and Na+ channels are in their deactivated state, blocked on the extracellular side by their activation gates. In response to an increase of the membrane potential to about −55 mV (in this case, caused by an action potential), the activation gates open, allowing positively charged Na+ ions to flow into the neuron through the channels, and causing the voltage across the neuronal membrane to increase to +30 mV in human neurons. Because the voltage across the membrane is initially negative, as its voltage increases to and past zero (from −70 mV at rest to a maximum of +30 mV), it is said to depolarize. This increase in voltage constitutes the rising phase of an action potential.
| Action Potential | Membrane Potential | Target Potential | Gate's Target State | Neuron's Target State |
|---|---|---|---|---|
| Resting | −70 mV | −55 mV | Deactivated → Activated | Polarized |
| Rising | −55 mV | 0 mV | Activated | Polarized → Depolarized |
| Rising | 0 mV | +30 mV | Activated → Inactivated | Depolarized |
| Falling | +30 mV | 0 mV | Inactivated | Depolarized → Repolarized |
| Falling | 0 mV | −70 mV | Inactivated | Repolarized |
| Undershot | −70 mV | −75 mV | Inactivated → Deactivated | Repolarized → Hyperpolarized |
| Rebounding | −75 mV | −70 mV | Deactivated | Hyperpolarized → Polarized |
At the peak of the action potential, when enough Na+ has entered the neuron and the membrane's potential has become high enough, the Na+ channels inactivate themselves by closing their inactivation gates. The inactivation gate can be thought of as a "plug" tethered to domains III and IV of the channel's intracellular alpha subunit. Closure of the inactivation gate causes Na+ flow through the channel to stop, which in turn causes the membrane potential to stop rising. The closing of the inactivation gate creates a refractory period within each individual Na+ channel. This refractory period eliminates the possibility of an action potential moving in the opposite direction back towards the soma. With its inactivation gate closed, the channel is said to be inactivated. With the Na+ channel no longer contributing to the membrane potential, the potential decreases back to its resting potential as the neuron repolarizes and subsequently hyperpolarizes itself, and this constitutes the falling phase of an action potential. The refractory period of each channel is therefore vital in propagating the action potential unidirectionally down an axon for proper communication between neurons.
When the membrane's voltage becomes low enough, the inactivation gate reopens and the activation gate closes in a process called deinactivation. With the activation gate closed and the inactivation gate open, the Na+ channel is once again in its deactivated state, and is ready to participate in another action potential.
When any kind of ion channel does not inactivate itself, it is said to be persistently (or tonically) active. Some kinds of ion channels are naturally persistently active. However, genetic mutations that cause persistent activity in other channels can cause disease by creating excessive activity of certain kinds of neurons. Mutations that interfere with Na+ channel inactivation can contribute to cardiovascular diseases or epileptic seizures by window currents, which can cause muscle and/or nerve cells to become over-excited.
Modeling the behavior of gates
The temporal behavior of Na+ channels can be modeled by a Markovian scheme or by the Hodgkin–Huxley-type formalism. In the former scheme, each channel occupies a distinct state with differential equations describing transitions between states; in the latter, the channels are treated as a population that are affected by three independent gating variables. Each of these variables can attain a value between 1 (fully permeant to ions) and 0 (fully non-permeant), the product of these variables yielding the percentage of conducting channels. The Hodgkin–Huxley model can be shown to be equivalent to a Markovian model (Explain|date=September 2021)
Impermeability to other ions
The pore of sodium channels contains a selectivity filter made of negatively charged amino acid residues, which attract the positive Na+ ion and keep out negatively charged ions such as chloride. The cations flow into a more constricted part of the pore that is 0.3 by 0.5 nm wide, which is just large enough to allow a single Na+ ion with a water molecule associated to pass through. The larger K+ ion cannot fit through this area. Ions of different sizes also cannot interact as well with the negatively charged glutamic acid residues that line the pore.
Diversity
Voltage-gated sodium channels normally consist of an alpha subunit that forms the ion conduction pore and one to two beta subunits that have several functions including modulation of channel gating.[6] Expression of the alpha subunit alone is sufficient to produce a functional channel.
Alpha subunits
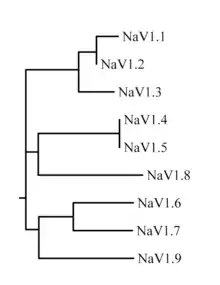
The family of sodium channels has nine known members, with amino acid identity >50% in the trans-membrane segments and extracellular loop regions. A standardized nomenclature for sodium channels is currently used and is maintained by the IUPHAR.[7][8]
The proteins of these channels are named Nav1.1 through Nav1.9. The gene names are referred to as SCN1A through SCN11A (the SCN6/7A gene is part of the Nax sub-family and has uncertain function). The likely evolutionary relationship between these channels, based on the similarity of their amino acid sequences, is shown in figure 1. The individual sodium channels are distinguished not only by differences in their sequence but also by their kinetics and expression profiles. Some of this data is summarized in table 1, below.
| Protein name | Gene | Expression profile | Associated human channelopathies |
|---|---|---|---|
| Nav1.1 | SCN1A | Central neurons, [peripheral neurons] and cardiac myocytes | febrile epilepsy, GEFS+, Dravet syndrome (also known as severe myclonic epilepsy of infancy or SMEI), borderline SMEI (SMEB), West syndrome (also known as infantile spasms), Doose syndrome (also known as myoclonic astatic epilepsy), intractable childhood epilepsy with generalized tonic-clonic seizures (ICEGTC), Panayiotopoulos syndrome, familial hemiplegic migraine (FHM), familial autism, Rasmussens's encephalitis and Lennox-Gastaut syndrome[9] |
| Nav1.2 | SCN2A | Central neurons, peripheral neurons | inherited febrile seizures, epilepsy, and autism spectrum disorder |
| Nav1.3 | SCN3A | Central neurons, peripheral neurons and cardiac myocytes | epilepsy, pain, brain malformations[10][11] |
| Nav1.4 | SCN4A | Skeletal muscle | hyperkalemic periodic paralysis, paramyotonia congenita, and potassium-aggravated myotonia |
| Nav1.5 | SCN5A | Cardiac myocytes, uninnervated skeletal muscle, central neurons, gastrointestinal smooth muscle cells and Interstitial cells of Cajal | Cardiac: Long QT syndrome Type 3, Brugada syndrome, progressive cardiac conduction disease, familial atrial fibrillation and idiopathic ventricular fibrillation;[12]
Gastrointestinal: Irritable bowel syndrome;[13] |
| Nav1.6 | SCN8A | Central neurons, dorsal root ganglia, peripheral neurons, heart, glia cells | Epilepsy,[14] ataxia, dystonia, tremor[15] |
| Nav1.7 | SCN9A | Dorsal root ganglia, sympathetic neurons, Schwann cells, and neuroendocrine cells | erythromelalgia, PEPD, channelopathy-associated insensitivity to pain[10] and recently discovered a disabling form of fibromyalgia (rs6754031 polymorphism)[16] |
| Nav1.8 | SCN10A | Dorsal root ganglia | pain,[10] neuropsychiatric disorders |
| Nav1.9 | SCN11A | Dorsal root ganglia | pain[10] |
| Nax | SCN7A | heart, uterus, skeletal muscle, astrocytes, dorsal root ganglion cells | none known |
Beta subunits
Sodium channel beta subunits are type 1 transmembrane glycoproteins with an extracellular N-terminus and a cytoplasmic C-terminus. As members of the Ig superfamily, beta subunits contain a prototypic V-set Ig loop in their extracellular domain. They do not share any homology with their counterparts of calcium and potassium channels.[17] Instead, they are homologous to neural cell adhesion molecules (CAMs) and the large family of L1 CAMs. There are four distinct betas named in order of discovery: SCN1B, SCN2B, SCN3B, SCN4B (table 2). Beta 1 and beta 3 interact with the alpha subunit non-covalently, whereas beta 2 and beta 4 associate with alpha via disulfide bond.[18] Sodium channels are more likely to stay open at the subthreshold membrane potential when interacting with beta toxins, which in turn induces an immediate sensation of pain.[19]
Role of beta subunits as cell adhesion molecules
In addition to regulating channel gating, sodium channel beta subunits also modulate channel expression and form links to the intracellular cytoskeleton via ankyrin and spectrin.[6][20][21] Voltage-gated sodium channels also assemble with a variety of other proteins, such as FHF proteins (Fibroblast growth factor Homologous Factor), calmodulin, cytoskeleton or regulatory kinases,[22][6][23][24][25] which form a complex with sodium channels, influencing its expression and/or function. Several beta subunits interact with one or more extracellular matrix (ECM) molecules. Contactin, also known as F3 or F11, associates with beta 1 as shown via co-immunoprecipitation.[26] Fibronectin-like (FN-like) repeats of Tenascin-C and Tenascin-R bind with beta 2 in contrast to the Epidermal growth factor-like (EGF-like) repeats that repel beta2.[27] A disintegrin and metalloproteinase (ADAM) 10 sheds beta 2's ectodomain possibly inducing neurite outgrowth.[28] Beta 3 and beta 1 bind to neurofascin at Nodes of Ranvier in developing neurons.[29]
| Protein name | Gene link | Assembles with | Expression profile | Associated human channelopathies |
|---|---|---|---|---|
| Navβ1 | SCN1B | Nav1.1 to Nav1.7 | Central Neurons, Peripheral Neurons, skeletal muscle, heart, glia | epilepsy (GEFS+), Brugada syndrome[30] |
| Navβ2 | SCN2B | Nav1.1, Nav1.2, Nav1.5 to Nav1.7 | Central Neurons, peripheral neurons, heart, glia | Brugada syndrome[30] |
| Navβ3 | SCN3B | Nav1.1 to Nav1.3, Nav1.5 | central neurons, adrenal gland, kidney, peripheral neurons | Brugada syndrome[30] |
| Navβ4 | SCN4B | Nav1.1, Nav1.2, Nav1.5 | heart, skeletal muscle, central and peripheral neurons | none known |
Ligand-gated sodium channels
Ligand-gated sodium channels are activated by binding of a ligand instead of a change in membrane potential.
They are found, e.g. in the neuromuscular junction as nicotinic receptors, where the ligands are acetylcholine molecules. Most channels of this type are permeable to potassium to some degree as well as to sodium.
Role in action potential
Voltage-gated sodium channels play an important role in action potentials. If enough channels open when there is a change in the cell's membrane potential, a small but significant number of Na+ ions will move into the cell down their electrochemical gradient, further depolarizing the cell. Thus, the more Na+ channels localized in a region of a cell's membrane the faster the action potential will propagate and the more excitable that area of the cell will be. This is an example of a positive feedback loop. The ability of these channels to assume a closed-inactivated state causes the refractory period and is critical for the propagation of action potentials down an axon.
Na+ channels both open and close more quickly than K+ channels, producing an influx of positive charge (Na+) toward the beginning of the action potential and an efflux (K+) toward the end.
Ligand-gated sodium channels, on the other hand, create the change in the membrane potential in the first place, in response to the binding of a ligand to it.
Pharmacologic modulation
Blockers
Activators
The following naturally produced substances persistently activate (open) sodium channels:
- Alkaloid-based toxins
- aconitine
- batrachotoxin
- brevetoxin
- ciguatoxin
- delphinine
- some grayanotoxins, e.g., grayanotoxin I (other granotoxins inactive, or close, sodium channels)
- veratridine
Gating modifiers
The following toxins modify the gating of sodium channels:
- Peptide-based toxins
- μ-Conotoxin
- δ-Atracotoxin[31]
- Scorpion venom toxins[32]
pH modulation
Changes in blood and tissue pH accompany physiological and pathophysiological conditions such as exercise, cardiac ischemia, ischemic stroke, and cocaine ingestion. These conditions are known to trigger the symptoms of electrical diseases in patients carrying sodium channel mutations. Protons cause a diverse set of changes to sodium channel gating, which generally lead to decreases in the amplitude of the transient sodium current and increases in the fraction of non-inactivating channels that pass persistent currents. These effects are shared with disease-causing mutants in neuronal, skeletal muscle, and cardiac tissue and may be compounded in mutants that impart greater proton sensitivity to sodium channels, suggesting a role of protons in triggering acute symptoms of electrical disease.[33]
Molecular mechanisms of proton block
Single channel data from cardiomyocytes have shown that protons can decrease the conductance of individual sodium channels.[34] The sodium channel selectivity filter is composed of a single residue in each of the four pore-loops of the four functional domains. These four residues are known as the DEKA motif.[35] The permeation rate of sodium through the sodium channel is determined by a four carboxylate residues, the EEDD motif, which make up the outer charged ring.[35] The protonation of these carboxylates is one of the main drivers of proton block in sodium channels, although there are other residues that also contribute to pH sensitivity.[36] One such residue is C373 in the cardiac sodium channel which makes it the most pH-sensitive sodium channel among the sodium channels that have been studied to date.[37]
pH modulation of sodium channel gating
As the cardiac sodium channel is the most pH-sensitive sodium channel, most of what is known is based on this channel. Reduction in extracellular pH has been shown to depolarize the voltage-dependence of activation and inactivation to more positive potentials. This indicates that during activities that decrease the blood pH, such as exercising, the probability of channels activating and inactivating is higher more positive membrane potentials, which can lead to potential adverse effects.[38] The sodium channels expressed in skeletal muscle fibers have evolved into relatively pH-insensitive channels. This has been suggested to be a protective mechanism against potential over- or under-excitability in skeletal muscles, as blood pH levels are highly susceptible to change during movement.[39][40] Recently, a mixed syndrome mutation that causes periodic paralysis and myotonia in the skeletal sodium channel has been shown to impart pH-sensitivity in this channel, making the gating of this channel similar to that of the cardiac subtype.[41]
pH modulation across the subtypes studied thus far
The effects of protonation have been characterized in Nav1.1-Nav1.5. Among these channels, Nav1.1-Nav1.3 and Nav1.5 display depolarized voltage-dependence of activation, while activation in Nav1.4 remains insensitive to acidosis. The voltage-dependence of steady-state fast inactivation is unchanged in Nav1.1-Nav1.4, but steady-state fast inactivation in Nav1.5 is depolarized. Hence, among the sodium channels that have been studied so far, Nav1.4 is the least and Nav1.5 is the most proton-sensitive subtypes.[42]
Evolution
A voltage-gated sodium channel is present in members of the choanoflagellates, thought to be the closest living, unicellular relative of animals.[43][44] This suggests that an ancestral form of the animal channel was among the many proteins that play central roles in animal life, but which are thought to have evolved before multicellularity.[45] The four-domain animal voltage-gated sodium channel likely evolved from a single-subunit ion channel, which was probably permeable for potassium ions, via a sequence of two duplication events.[46] This model draws support from the fact that subunits I and III (and II and IV) group by similarity, suggesting that a two-channel intermediate generated from the first duplication existed long enough for divergence to occur between its two subunits. After the second duplication, the channel was left with two sets of similar domains.[46] The resulting four-domain channel is thought to have been permeable primarily for calcium, and to have achieved sodium selectivity a number of times independently.[47][48] After divergence from the invertebrates, the vertebrate lineage underwent two whole-genome duplications (WGDs), yielding a set of four sodium channel gene prologues in the ancestral vertebrate, all of which were retained.[49][50] After the tetrapod/teleost split, the teleosts likely underwent a third WGD leading to the eight sodium channel prologues expressed in many modern fishes.[49] The modern, ten-paralogue sodium gene complement of mammals is thought to have arisen from a series of parallel and nested duplications involving two of the four paralogues present in the ancestor of all tetrapods.[50]
See also
- Calcium channel – Ion channel complex through which calcium ions pass
- Chronaxie – Electrophysiology metric
- Epithelial sodium channel – Group of membrane proteins
- Ion channel – Pore-forming membrane protein
- Resting ion channel
- Sodium in biology – Use of Sodium by organisms
References
- Jessell TM, Kandel ER, Schwartz JH (2000). Principles of Neural Science (4th ed.). New York: McGraw-Hill. pp. 154–69. ISBN 978-0-8385-7701-1.
{{cite book}}: CS1 maint: multiple names: authors list (link) - Bertil Hillel (2001). Ion Channels of Excitable Membranes (3rd ed.). Sunderland, Mass: Sinauer. pp. 73–7. ISBN 978-0-87893-321-1.
- Lim C, Dudev T (2016). "Chapter 10. Potassium Versus Sodium Selectivity in Monovalent Ion Channel Selectivity Filters". In Astrid S, Helmut S, Roland KO S (eds.). The Alkali Metal Ions: Their Role in Life. Metal Ions in Life Sciences. Vol. 16. Springer. pp. 325–347. doi:10.1007/978-3-319-21756-7_9. PMID 26860305.
- Yu FH, Catterall WA (2003). "Overview of the voltage-gated sodium channel family". Genome Biology. 4 (3): 207. doi:10.1186/gb-2003-4-3-207. PMC 153452. PMID 12620097.
- Nicholls, Martin, Fuchs, Brown, Diamond, Weisblat. (2012) "From Neuron to Brain," 5th ed. pg. 86
- Isom LL (February 2001). "Sodium channel beta subunits: anything but auxiliary". The Neuroscientist. 7 (1): 42–54. doi:10.1177/107385840100700108. PMID 11486343. S2CID 86422657.
- IUPHAR – International Union of Basic and Clinical Pharmacology
- Catterall WA, Goldin AL, Waxman SG (December 2005). "International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels". Pharmacological Reviews. 57 (4): 397–409. doi:10.1124/pr.57.4.4. PMID 16382098. S2CID 7332624.
- Lossin C. "SCN1A infobase". Archived from the original on 2011-07-21. Retrieved 2009-10-30.
compilation of genetic variations in the SCN1A gene that alter the expression or function of Nav1.1
- Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD (April 2019). "The Role of Voltage-Gated Sodium Channels in Pain Signaling". Physiological Reviews. 99 (2): 1079–1151. doi:10.1152/physrev.00052.2017. PMID 30672368.
- Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, et al. (September 2018). "V1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development". Neuron. 99 (5): 905–913.e7. doi:10.1016/j.neuron.2018.07.052. PMC 6226006. PMID 30146301.
- Chockalingam P, Wilde A (September 2012). "The multifaceted cardiac sodium channel and its clinical implications". Heart. 98 (17): 1318–24. doi:10.1136/heartjnl-2012-301784. PMID 22875823. S2CID 44433455.
- Beyder A, Mazzone A, Strege PR, Tester DJ, Saito YA, Bernard CE, Enders FT, Ek WE, Schmidt PT, Dlugosz A, Lindberg G, Karling P, Ohlsson B, Gazouli M, Nardone G, Cuomo R, Usai-Satta P, Galeazzi F, Neri M, Portincasa P, Bellini M, Barbara G, Camilleri M, Locke GR, Talley NJ, D'Amato M, Ackerman MJ, Farrugia G (June 2014). "Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome". Gastroenterology. 146 (7): 1659–1668. doi:10.1053/j.gastro.2014.02.054. PMC 4096335. PMID 24613995.
- Butler KM, da Silva C, Shafir Y, Weisfeld-Adams JD, Alexander JJ, Hegde M, Escayg A (January 2017). "De novo and inherited SCN8A epilepsy mutations detected by gene panel analysis". Epilepsy Research. 129: 17–25. doi:10.1016/j.eplepsyres.2016.11.002. PMC 5321682. PMID 27875746.
- Meisler MH, Kearney JA (August 2005). "Sodium channel mutations in epilepsy and other neurological disorders". The Journal of Clinical Investigation. 115 (8): 2010–7. doi:10.1172/JCI25466. PMC 1180547. PMID 16075041.
- Vargas-Alarcon G, Alvarez-Leon E, Fragoso JM, Vargas A, Martinez A, Vallejo M, Martinez-Lavin M (February 2012). "A SCN9A gene-encoded dorsal root ganglia sodium channel polymorphism associated with severe fibromyalgia". BMC Musculoskeletal Disorders. 13: 23. doi:10.1186/1471-2474-13-23. PMC 3310736. PMID 22348792.
- Catterall WA (April 2000). "From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels". Neuron. 26 (1): 13–25. doi:10.1016/S0896-6273(00)81133-2. PMID 10798388. S2CID 17928749.
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (May 1992). "Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel". Science. 256 (5058): 839–42. Bibcode:1992Sci...256..839I. doi:10.1126/science.1375395. PMID 1375395.
- blackboard.jhu.edu (PDF) https://blackboard.jhu.edu/courses/1/AS.410.603.83.SU20/db/_10095565_1/Isolation%20and%20Characterization%20of%20CvIV4%20A%20Pain%20Inducing%20a-scorpion%20Toxin.pdf. Retrieved 2020-07-16.
{{cite web}}: Missing or empty|title=(help) - Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL (April 2000). "Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact". The Journal of Biological Chemistry. 275 (15): 11383–8. doi:10.1074/jbc.275.15.11383. PMID 10753953.
- Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL (July 2002). "Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin". The Journal of Biological Chemistry. 277 (29): 26681–8. doi:10.1074/jbc.M202354200. PMID 11997395.
- Cantrell AR, Catterall WA (June 2001). "Neuromodulation of Na+ channels: an unexpected form of cellular plasticity". Nature Reviews. Neuroscience. 2 (6): 397–407. doi:10.1038/35077553. PMID 11389473. S2CID 22885909.
- Shah BS, Rush AM, Liu S, Tyrrell L, Black JA, Dib-Hajj SD, Waxman SG (August 2004). "Contactin associates with sodium channel Nav1.3 in native tissues and increases channel density at the cell surface". The Journal of Neuroscience. 24 (33): 7387–99. doi:10.1523/JNEUROSCI.0322-04.2004. PMC 6729770. PMID 15317864.
- Wittmack EK, Rush AM, Craner MJ, Goldfarb M, Waxman SG, Dib-Hajj SD (July 2004). "Fibroblast growth factor homologous factor 2B: association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons". The Journal of Neuroscience. 24 (30): 6765–75. doi:10.1523/JNEUROSCI.1628-04.2004. PMC 6729706. PMID 15282281.
- Rush AM, Wittmack EK, Tyrrell L, Black JA, Dib-Hajj SD, Waxman SG (May 2006). "Differential modulation of sodium channel Na(v)1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily". The European Journal of Neuroscience. 23 (10): 2551–62. doi:10.1111/j.1460-9568.2006.04789.x. PMID 16817858. S2CID 21411801.
- Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao ZC (October 2001). "Contactin associates with Na+ channels and increases their functional expression". The Journal of Neuroscience. 21 (19): 7517–25. doi:10.1523/JNEUROSCI.21-19-07517.2001. PMC 6762905. PMID 11567041.
- Srinivasan J, Schachner M, Catterall WA (December 1998). "Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R". Proceedings of the National Academy of Sciences of the United States of America. 95 (26): 15753–7. Bibcode:1998PNAS...9515753S. doi:10.1073/pnas.95.26.15753. PMC 28116. PMID 9861042.
- Kim DY, Ingano LA, Carey BW, Pettingell WH, Kovacs DM (June 2005). "Presenilin/gamma-secretase-mediated cleavage of the voltage-gated sodium channel beta2-subunit regulates cell adhesion and migration". The Journal of Biological Chemistry. 280 (24): 23251–61. doi:10.1074/jbc.M412938200. PMID 15833746.
- Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA (July 2001). "Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain". The Journal of Cell Biology. 154 (2): 427–34. doi:10.1083/jcb.200102086. PMC 2150779. PMID 11470829.
- Antzelevitch C, Patocskai B (January 2016). "Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects". Current Problems in Cardiology. 41 (1): 7–57. doi:10.1016/j.cpcardiol.2015.06.002. PMC 4737702. PMID 26671757.
- Grolleau F, Stankiewicz M, Birinyi-Strachan L, Wang XH, Nicholson GM, Pelhate M, Lapied B (February 2001). "Electrophysiological analysis of the neurotoxic action of a funnel-web spider toxin, delta-atracotoxin-HV1a, on insect voltage-gated Na+ channels". The Journal of Experimental Biology. 204 (Pt 4): 711–21. doi:10.1242/jeb.204.4.711. PMID 11171353.
- Possani LD, Becerril B, Delepierre M, Tytgat J (September 1999). "Scorpion toxins specific for Na+-channels". European Journal of Biochemistry. 264 (2): 287–300. doi:10.1046/j.1432-1327.1999.00625.x. PMID 10491073.
- Peters CH, Ghovanloo MR, Gershome C, Ruben PC (February 2018). "pH Modulation of Voltage-Gated Sodium Channels". Voltage-gated Sodium Channels: Structure, Function and Channelopathies. Handbook of Experimental Pharmacology. Vol. 246. pp. 147–160. doi:10.1007/164_2018_99. ISBN 978-3-319-90283-8. PMID 29460150.
- Zhang JF, Siegelbaum SA (December 1991). "Effects of external protons on single cardiac sodium channels from guinea pig ventricular myocytes". The Journal of General Physiology. 98 (6): 1065–83. doi:10.1085/jgp.98.6.1065. PMC 2229074. PMID 1664454.
- Sun YM, Favre I, Schild L, Moczydlowski E (December 1997). "On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel. Effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving". The Journal of General Physiology. 110 (6): 693–715. doi:10.1085/jgp.110.6.693. PMC 2229404. PMID 9382897.
- Khan A, Romantseva L, Lam A, Lipkind G, Fozzard HA (August 2002). "Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block". The Journal of Physiology. 543 (Pt 1): 71–84. doi:10.1113/jphysiol.2002.021014. PMC 2290475. PMID 12181282.
- Vilin YY, Peters CH, Ruben PC (2012). "Acidosis differentially modulates inactivation in na(v)1.2, na(v)1.4, and na(v)1.5 channels". Frontiers in Pharmacology. 3: 109. doi:10.3389/fphar.2012.00109. PMC 3372088. PMID 22701426.
- Jones DK, Peters CH, Allard CR, Claydon TW, Ruben PC (February 2013). "Proton sensors in the pore domain of the cardiac voltage-gated sodium channel". The Journal of Biological Chemistry. 288 (7): 4782–91. doi:10.1074/jbc.M112.434266. PMC 3576083. PMID 23283979.
- Khan A, Kyle JW, Hanck DA, Lipkind GM, Fozzard HA (October 2006). "Isoform-dependent interaction of voltage-gated sodium channels with protons". The Journal of Physiology. 576 (Pt 2): 493–501. doi:10.1113/jphysiol.2006.115659. PMC 1890365. PMID 16873405.
- Hermansen L, Osnes JB (March 1972). "Blood and muscle pH after maximal exercise in man". Journal of Applied Physiology. 32 (3): 304–8. doi:10.1152/jappl.1972.32.3.304. PMID 5010039.
- Ghovanloo MR, Abdelsayed M, Peters CH, Ruben PC (April 2018). "A Mixed Periodic Paralysis & Myotonia Mutant, P1158S, Imparts pH-Sensitivity in Skeletal Muscle Voltage-gated Sodium Channels". Scientific Reports. 8 (1): 6304. Bibcode:2018NatSR...8.6304G. doi:10.1038/s41598-018-24719-y. PMC 5908869. PMID 29674667.
- Ghovanloo MR, Peters CH, Ruben PC (October 2018). "Effects of acidosis on neuronal voltage-gated sodium channels: Nav1.1 and Nav1.3". Channels. 12 (1): 367–377. doi:10.1080/19336950.2018.1539611. PMC 6284583. PMID 30362397.
- Moran Y, Barzilai MG, Liebeskind BJ, Zakon HH (February 2015). "Evolution of voltage-gated ion channels at the emergence of Metazoa". The Journal of Experimental Biology. 218 (Pt 4): 515–25. doi:10.1242/jeb.110270. PMID 25696815.
- Liebeskind BJ, Hillis DM, Zakon HH (May 2011). "Evolution of sodium channels predates the origin of nervous systems in animals". Proceedings of the National Academy of Sciences of the United States of America. 108 (22): 9154–9. Bibcode:2011PNAS..108.9154L. doi:10.1073/pnas.1106363108. PMC 3107268. PMID 21576472.
- King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, et al. (February 2008). "The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans". Nature. 451 (7180): 783–8. Bibcode:2008Natur.451..783K. doi:10.1038/nature06617. PMC 2562698. PMID 18273011.
- Strong M, Chandy KG, Gutman GA (January 1993). "Molecular evolution of voltage-sensitive ion channel genes: on the origins of electrical excitability". Molecular Biology and Evolution. 10 (1): 221–42. doi:10.1093/oxfordjournals.molbev.a039986. PMID 7680747.
- Liebeskind BJ, Hillis DM, Zakon HH (November 2013). "Independent acquisition of sodium selectivity in bacterial and animal sodium channels". Current Biology. 23 (21): R948–9. doi:10.1016/j.cub.2013.09.025. PMID 24200318.
- Kasimova MA, Granata D, Carnevale V (2016). Voltage-Gated Sodium Channels: Evolutionary History and Distinctive Sequence Features. Current Topics in Membranes. Vol. 78. pp. 261–86. doi:10.1016/bs.ctm.2016.05.002. ISBN 9780128053867. PMID 27586287.
- Widmark J, Sundström G, Ocampo Daza D, Larhammar D (January 2011). "Differential evolution of voltage-gated sodium channels in tetrapods and teleost fishes". Molecular Biology and Evolution. 28 (1): 859–71. doi:10.1093/molbev/msq257. PMID 20924084.
- Zakon HH, Jost MC, Lu Y (April 2011). "Expansion of voltage-dependent Na+ channel gene family in early tetrapods coincided with the emergence of terrestriality and increased brain complexity". Molecular Biology and Evolution. 28 (4): 1415–24. doi:10.1093/molbev/msq325. PMC 3058772. PMID 21148285.
External links
- Sodium+Channels at the US National Library of Medicine Medical Subject Headings (MeSH)
- "Voltage-Gated Sodium Channels". IUPHAR Database of Receptors and Ion Channels. International Union of Basic and Clinical Pharmacology.