Lobeglitazone
 | |
| Clinical data | |
|---|---|
| Trade names | Duvie |
| Other names | CKD-501 |
| Routes of administration | Oral |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | liver (CYP2C9, 2C19, and 1A2)[1] |
| Elimination half-life | 7.8–9.8 hours[2] |
| Identifiers | |
IUPAC name
| |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
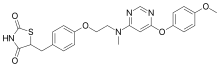
| Formula | C24H24N4O5S |
| Molar mass | 480.54 g·mol−1 |
| 3D model (JSmol) | |
SMILES
| |
InChI
| |
Lobeglitazone (trade name Duvie, Chong Kun Dang) is an antidiabetic drug in the thiazolidinedione class of drugs. As an agonist for both PPARα and PPARγ, it works as an insulin sensitizer by binding to the PPAR receptors in fat cells and making the cells more responsive to insulin.[3]
Medical uses
Lobeglitazone is used to assist regulation of blood glucose level of diabetes mellitus type 2 patients. It can be used alone or in combination with metformin.[4]
Lobeglitazone was approved by the Ministry of Food and Drug Safety (Korea) in 2013, and the postmarketing surveillance is on progress until 2019.[4][5]
Pharmacokinetics
The absolute bioavailability of lobeglitazone is about 95% in rat.[1] In human, the mean steady state clearance (CLss/F) was 1.13 L/h across in 1 to 4 mg dose range. In the dose range, the mean half-life was 10.3 h.[2] Urine excretion was negligible amount in elimination of lobeglitazone in rat and human.[1][2]
The plasma protein binding of the drug is over 99%.[1][6] The average blood-to-plasma concentration ratio was 0.636. The unbound fraction of lobeglitazone in microsomal incubation medium was 0.479.[6]
Lobeglitazone was primarily distributed to the liver with tissue-to-plasma concentration ratio as 5.59, and less to heart, lung, and fat. The tissue to plasma concentration ratios were ranged from about 0.25 to 4.0 for major tissues, in rat.[1]
Among six major membrane transporters recommended by the United States Food and Drug Administration, lobeglitazone interacts with OATP1B1, OAT3, and MDR1.[1] In vitro, lobeglitazone was a substrate of rodent OATP1B2.[6] Lobeglitazone interacted with CYP1A2, 2C9 and 2C19.[1]
Distribution to liver of lobeglitazone was inhibited by atorvastatin, in rats.[6]
References
- 1 2 3 4 5 6 7 8 Lee JH, Noh CK, Yim CS, Jeong YS, Ahn SH, Lee W, Kim DD, Chung SJ (2015). "Kinetics of the Absorption, Distribution, Metabolism, and Excretion of Lobeglitazone, a Novel Activator of Peroxisome Proliferator-Activated Receptor Gamma in Rats". Journal of Pharmaceutical Sciences. 104 (9): 3049–3059. doi:10.1002/jps.24378. PMID 25648999.
- 1 2 3 Kim JW, Kim JR, Yi S, Shin KH, Shin HS, Yoon SH, Cho JY, Kim DH, Shin SG, Jang IJ, Yu KS (2011). "Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-γ agonist: a single- and multiple-dose, double-blind, randomized control study in healthy male Korean subjects". Clinical Therapeutics. 33 (11): 1819–1830. doi:10.1016/j.clinthera.2011.09.023. PMID 22047812.
- ↑ Lee JH, Woo YA, Hwang IC, Kim CY, Kim DD, Shim CK, Chung SJ (2009). "Quantification of CKD-501, lobeglitazone, in rat plasma using a liquid-chromatography/tandem mass spectrometry method and its applications to pharmacokinetic studies". Journal of Pharmaceutical and Biomedical Analysis. 50 (5): 872–877. doi:10.1016/j.jpba.2009.06.003. PMID 19577404.
- 1 2 "MFDS permission information of Duvie Tablet 0.5mg". Ministry of Food and Drug Safety. Archived from the original (Release of Information) on 3 March 2016. Retrieved 23 October 2014.
- ↑ "국내개발 20번째 신약'듀비에정'허가(20th new drug developed in Korea 'Duvie Tablet' was approved)". Chong Kun Dang press release. 4 July 2013. Archived from the original on 23 September 2015. Retrieved 23 October 2014.
- 1 2 3 4 Yim CS, Jeong YS, Lee SY, Pyeon W, Ryu HM, Lee JH, Lee KR, Maeng HJ, Chung SJ (2017). "Specific Inhibition of the Distribution of Lobeglitazone to the Liver by Atorvastatin in Rats: Evidence for an rOATP1B2-Mediated Interaction in Hepatic Transport". Drug Metabolism and Disposition. 45 (3): 246–259. doi:10.1124/dmd.116.074120. PMID 28069721.