Glutamate receptor
Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells.[1] Glutamate (the conjugate base of glutamic acid) is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter.[2] Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
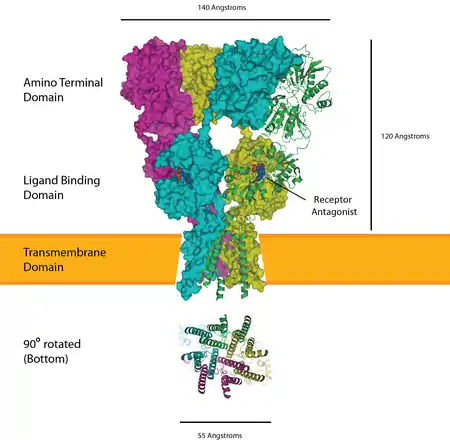
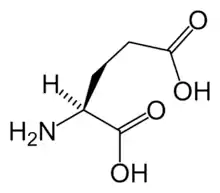
Glutamate receptors are implicated in a number of neurological conditions. Their central role in excitotoxicity and prevalence in the central nervous system has been linked or speculated to be linked to many neurodegenerative diseases, and several other conditions have been further linked to glutamate receptor gene mutations or receptor autoantigen/antibody activity.
Function
Glutamate
Glutamate is the most prominent neurotransmitter in the body, and is the main excitatory neurotransmitter, being present in over 50% of nervous tissue.[2][3] Glutamate was initially discovered to be a neurotransmitter in insect studies in the early 1960s.
Glutamate is also used by the brain to synthesize GABA (γ-Aminobutyric acid), the main inhibitory neurotransmitter of the mammalian central nervous system. GABA plays a role in regulating neuronal excitability throughout the nervous system and is also directly responsible for the regulation of muscle tone in humans.[2][4]
Glutamate receptors
Mammalian glutamate receptors are classified based on their pharmacology.[5] However, glutamate receptors in other organisms have different pharmacology, and therefore these classifications do not hold. One of the major functions of glutamate receptors appears to be the modulation of synaptic plasticity, a property of the brain thought to be vital for memory and learning. Both metabotropic and ionotropic glutamate receptors have been shown to have an effect on synaptic plasticity.[6] An increase or decrease in the number of ionotropic glutamate receptors on a postsynaptic cell may lead to long-term potentiation or long-term depression of that cell, respectively.[7][8][9] Additionally, metabotropic glutamate receptors may modulate synaptic plasticity by regulating postsynaptic protein synthesis through second messenger systems.[10] Research shows that glutamate receptors are present in CNS glial cells as well as neurons.[11] These glutamate receptors are suggested to play a role in modulating gene expression in glial cells, both during the proliferation and differentiation of glial precursor cells in brain development and in mature glial cells.[12]
Types
Ionotropic glutamate receptors (iGluRs) form the ion channel pore that activates when glutamate binds to the receptor. Metabotropic glutamate receptors (mGluRs) affect the cell through a signal transduction cascade, and they may be primarily activating (mGlur1/5) or primarily inhibitory (mGlur2/3 and mGlur4/6/7/8). Ionotropic receptors tend to be quicker in relaying information, but metabotropic ones are associated with a more prolonged stimulus. The signalling cascade induced by metabotropic receptor activation means that even a relatively brief or small synaptic signal can have large and long-lasting effects, i.e. the system can have high "gain." NMDA receptor activation is particularly complex, as channel opening requires not only glutamate binding but also glycine or serine binding simultaneously at a separate site, and it also displays a degree of voltage dependence due to Zn2+ or Mg2+ binding in the pore.[13] Furthermore, Ca2+ currents through the NMDA receptor modulate not just the membrane potential but act as an important second messenger system. The particular dynamics of the NMDAR allow it to function as a neural coincidence detector, and the NMDAR Ca2+ currents are critical in synaptic plasticity (LTP and LTD) and learning and memory in general.[13]
Of the many specific subtypes of glutamate receptors, it is customary to refer to primary subtypes by a chemical that binds to it more selectively than glutamate. The research, however, is ongoing, as subtypes are identified and chemical affinities measured. Several compounds are routinely used in glutamate receptor research and associated with receptor subtypes:
| Type | Name | Agonist(s) | Antagonists |
| ionotropic | NMDA receptor | NMDA | Ketamine |
| Kainate receptor | Kainate | UBP-302 | |
| AMPA receptor | AMPA | Perampanel | |
| Group 1 metabotropic | mGluR1, mGluR5 | DHPG | LY-344,545 |
| Group 2 metabotropic | mGluR2, mGluR3 | DCG-IV | LY-341,495 |
| Group 3 metabotropic | mGluR4, mGluR6, mGluR7, mGluR8 | L-AP4 | MMPIP (mGlur7) |
Due to the diversity of glutamate receptors, their subunits are encoded by numerous gene families. Sequence similarities between mammals show a common evolutionary origin for many mGluR and all iGluR genes.[14] Conservation of reading frames and splice sites of GluR genes between chimpanzees and humans is complete, suggesting no gross structural changes after humans diverged from the human-chimpanzee common ancestor. However, there is a possibility that two human-specific "fixed" amino acid substitutions, D71G in GRIN3A and R727H in GRIN3B, are specifically associated with human brain function.[15]
Ionotropic
Mammalian ionotropic glutamate receptor subunits and their genes:[16][17]
| Mammalian receptor family | Subunit
(Old nomenclature) |
Gene | Chromosome (human) |
|---|---|---|---|
| AMPA | GluA1 (GluR1) | GRIA1 | 5q33 |
| GluA2 (GluR2) | GRIA2 | 4q32-33 | |
| GluA3 (GluR3) | GRIA3 | Xq25-26 | |
| GluA4 (GluR4) | GRIA4 | 11q22-23 | |
| Kainate | GluK1 (GluR5) | GRIK1 | 21q21.1-22.1 |
| GluK2 (GluR6) | GRIK2 | 6q16.3-q21 | |
| GluK3 (GluR7) | GRIK3 | 1p34-p33 | |
| GluK4 (KA-1) | GRIK4 | 11q22.3 | |
| GluK5 (KA-2) | GRIK5 | 19q13.2 | |
| NMDA | GluN1(NR1) | GRIN1 | 9q34.3 |
| GluN2A (NR2A) | GRIN2A | 16p13.2 | |
| GluN2B (NR2B) | GRIN2B | 12p12 | |
| GluN2C (NR2C) | GRIN2C | 17q24-q25 | |
| GluN2D (NR2D) | GRIN2D | 19q13.1qter | |
| GluN3A (NR3A) | GRIN3A | 9q31.1 | |
| GluN3B (NR3B) | GRIN3B | 19p13.3 |
Metabotropic
Mammalian metabotropic glutamate receptors are all named mGluR# and are further broken down into three groups:
| Group | Receptor | Gene | Chromosome (human) |
Effect |
|---|---|---|---|---|
| 1 | mGluR1 | GRM1 | 6q24 | Increase in Ca2+ concentration in the cytoplasm. |
| mGluR5 | GRM5 | 11q14.3 | Release of K+ from the cell by activating K+ ionic channels | |
| 2 | mGluR2 | GRM2 | 3p21.2 | Inhibition of adenylyl cyclase causing shutdown of the cAMP-dependent pathway And therefore decreasing amount of cAMP |
| mGluR3 | GRM3 | 7q21.1-q21.2 | ||
| 3 | mGluR4 | GRM4 | 6p21.3 | Activation of Ca2+ channels, allowing more Ca2+ to enter the cell[18] |
| mGluR6 | GRM6 | 5q35 | ||
| mGluR7 | GRM7 | 3p26-p25 | ||
| mGluR8 | GRM8 | 7q31.3-q32.1 |
In other (non mammalian) organisms, the classification and subunit composition of glutamate receptors is different.
Structure, mechanism and function
Glutamate receptors exist primarily in the central nervous system. These receptors can be found on the dendrites of postsynaptic cells and bind to glutamate released into the synaptic cleft by presynaptic cells. They are also present on both astrocytes and oligodendrocytes.[12] Ionotropic and metabotropic glutamate receptors, with the exception of NMDA, are found on cultured glial cells, which can open in response to glutamate and cause cells to activate second messengers to regulate gene expression and release neuroactive compounds.[11] Furthermore, brain slices show glutamate receptors are ubiquitously expressed in both developing and mature astrocytes and oligodendrocytes in vivo. Because of this, glial glutamate receptors are thought to be vital for glial cell development.[12]
Ionotropic
Ionotropic glutamate receptors, by definition, are ligand-gated nonselective cation channels that allow the flow of K+, Na+ and sometimes Ca2+ in response to glutamate binding. (In C. elegans and Drosophila, invertebrate-specific subunits enable the flow of negative chloride ions rather than cations.) Upon binding, the agonist will stimulate direct action of the central pore of the receptor, an ion channel, allowing ion flow and causing excitatory postsynaptic current (EPSC). This current is depolarizing and, if enough glutamate receptors are activated, may trigger an action potential in the postsynaptic neuron. All produce excitatory postsynaptic current, but the speed and duration of the current is different for each type. NMDA receptors have an internal binding site for an Mg2+ ion, creating a voltage-dependent block, which is removed by outward flow of positive current.[19] Since the block must be removed by outward current flow, NMDA receptors rely on the EPSC produced by AMPA receptors to open. NMDA receptors are permeable to Ca2+,[20] which is an important cation in the nervous system[21] and has been linked to gene regulation.[22] The flow of Ca2+ through NMDA receptors is thought to cause both long-term potentiation (LTP, of synapse efficacy) and long-term depression (LTD) by transducing signaling cascades and regulating gene expression.
Metabotropic
Metabotropic glutamate receptors, which belong to subfamily C of G protein-coupled receptors are divided into three groups, with a total of eight subtypes (in mammals; this is not necessarily the case for most organisms). The mGluRs are composed of three distinct regions: the extracellular region, the transmembrane region, and the intracellular region.[23] The extracellular region is composed of a venus flytrap (VFT) module that binds glutamate,[24] and a cysteine-rich domain that is thought to play a role in transmitting the conformational change induced by ligand binding from in the VFT module to the transmembrane region.[23] The transmembrane region consists of seven transmembrane domains and connects the extracellular region to the intracellular region where G protein coupling occurs.[24] Glutamate binding to the extracellular region of an mGluR causes G proteins bound to the intracellular region to be phosphorylated, affecting multiple biochemical pathways and ion channels in the cell.[25] Because of this, mGluRs can both increase or decrease the excitability of the postsynaptic cell, thereby causing a wide range of physiological effects.
Outside the central nervous system
Glutamate receptors are thought to be responsible for the reception and transduction of umami taste stimuli. Taste receptors of the T1R family, belonging to the same class of GPCR as metabotropic glutamate receptors are involved. Additionally, the mGluRs, as well as ionotropic glutamate receptors in neural cells, have been found in taste buds and may contribute to the umami taste.[26] Numerous ionotropic glutamate receptor subunits are expressed by heart tissue, but their specific function is still unknown. Western blots and northern blots confirmed the presence of iGluRs in cardiac tissue. Immunohistochemistry localized the iGluRs to cardiac nerve terminals, ganglia, conducting fibers, and some myocardiocytes.[27] Glutamate receptors are (as mentioned above) also expressed in pancreatic islet cells.[28] AMPA iGluRs modulate the secretion of insulin and glucagon in the pancreas, opening the possibility of treatment of diabetes via glutamate receptor antagonists.[29][30] Small unmyelinated sensory nerve terminals in the skin also express NMDA and non-NMDA receptors. Subcutaneous injections of receptor blockers in rats successfully analgesized skin from formalin-induced inflammation, raising possibilities of targeting peripheral glutamate receptors in the skin for pain treatment.[31]
General clinical implications
Specific medical conditions and symptoms are discussed below.
Autoimmunity and antibody interactions with glutamate receptors and their subunit genes
Various neurological disorders are accompanied by antibody or autoantigen activity associated with glutamate receptors or their subunit genes (e.g. GluR3 in Rasmussen's encephalitis,[32] and GluR2 in nonfamilial olivopontocerebellar degeneration).[33] In 1994 GluR3 was shown to act as an autoantigen in Rasmussen's encephalitis, leading to speculation that autoimmune activity might underlie the condition.[34] Such findings "suggest" links between glutamate receptors and autoimmune interactions are possible and may be significant in some degenerative diseases,[33] however the exact role of such antibodies in disease manifestation is still not entirely known.[35]
Excitotoxicity
Overstimulation of glutamate receptors causes neurodegeneration and neuronal damage through a process called excitotoxicity. Excessive glutamate, or excitotoxins acting on the same glutamate receptors, overactivate glutamate receptors (specifically NMDARs), causing high levels of calcium ions (Ca2+) to influx into the postsynaptic cell.[36]
High Ca2+ concentrations activate a cascade of cell degradation processes involving proteases, lipases, nitric oxide synthase, and a number of enzymes that damage cell structures often to the point of cell death.[37] Ingestion of or exposure to excitotoxins that act on glutamate receptors can induce excitotoxicity and cause toxic effects on the central nervous system.[38] This becomes a problem for cells, as it feeds into a cycle of positive feedback cell death.
Glutamate excitotoxicity triggered by overstimulation of glutamate receptors also contributes to intracellular oxidative stress. Proximal glial cells use a cystine/glutamate antiporter (xCT) to transport cystine into the cell and glutamate out. Excessive extracellular glutamate concentrations reverse xCT, so glial cells no longer have enough cystine to synthesize glutathione (GSH), an antioxidant.[39] Lack of GSH leads to more reactive oxygen species (ROSs) that damage and kill the glial cell, which then cannot reuptake and process extracellular glutamate.[40] This is another positive feedback in glutamate excitotoxicity. In addition, increased Ca2+ concentrations activate nitric oxide synthase (NOS) and the over-synthesis of nitric oxide (NO). High NO concentration damages mitochondria, leading to more energy depletion, and adds oxidative stress to the neuron as NO is a ROS.[41]
Neurodegeneration
In the case of traumatic brain injury or cerebral ischemia (e.g., cerebral infarction or hemorrhage), acute neurodegeneration caused by excitotoxicity may spread to proximal neurons through two processes. Hypoxia and hypoglycemia trigger bioenergetic failure; mitochondria stop producing ATP energy. Na+/K+-ATPase can no longer maintain sodium/potassium ion concentration gradients across the plasma membrane. Glutamate transporters (EAATs), which use the Na+/K+ gradient, reverse glutamate transport (efflux) in affected neurons and astrocytes, and depolarization increases downstream synaptic release of glutamate.[42] In addition, cell death via lysis or apoptosis releases cytoplasmic glutamate outside of the ruptured cell.[43] These two forms of glutamate release cause a continual cascade of excitotoxic cell death and further increased extracellular glutamate concentrations.
Glutamate receptors' significance in excitotoxicity also links it to many neurogenerative diseases. Conditions such as exposure to excitotoxins, old age, congenital predisposition, and brain trauma can trigger glutamate receptor activation and ensuing excitotoxic neurodegeneration. This damage to the central nervous system propagates symptoms associated with a number of diseases.[44]
Conditions with demonstrated associations to glutamate receptors
A number of diseases in humans have a proven association with genetic mutations of glutamate receptor genes, or autoantigen/antibody interactions with glutamate receptors or their genes. Glutamate receptors and impaired regulation (in particular, those resulting in excessive glutamate levels) are also one cause of excitotoxicity (described above), which itself has been implicated or associated with a number of specific neurodegenerative conditions where neural cell death or degradation within the brain occurs over time.[40][44]
Excessive synaptic receptor stimulation by glutamate is directly related to many conditions. Magnesium is one of many antagonists at the glutamate receptor, and magnesium deficiencies have demonstrated relationships with many glutamate receptor-related conditions.[45]
Glutamate receptors have been found to have an influence in ischemia/stroke, seizures, Parkinson's disease, Huntington's disease, and aching,[46] addiction[47] and an association with both ADHD[48] and autism.[49]
In most cases these are areas of ongoing research.
Aching
Hyperalgesia is directly involved with spinal NMDA receptors. Administered NMDA antagonists in a clinical setting produce significant side effects, although more research is being done in intrathecal administration.[38] Since spinal NMDA receptors link the area of pain to the brain's pain processing center, the thalamus, these glutamate receptors are a prime target for treatment. One proposed way to cope with the pain is subconsciously through the visualization technique.[50]
Attention deficit hyperactivity disorder (ADHD)
In 2006 the glutamate receptor subunit gene GRIN2B (responsible for key functions in memory and learning) was associated with ADHD.[51] This followed earlier studies showing a link between glutamate modulation and hyperactivity (2001),[52] and then between the SLC1A3 solute carrier gene-encoding part of the glutamate transporter process that mapped to chromosome 5 (5p12) noted in multiple ADHD genome scans.[53]
Further mutations to four different metabotropic glutamate receptor genes were identified in a study of 1013 children with ADHD compared to 4105 controls with non-ADHD, replicated in a subsequent study of 2500 more patients. Deletions and duplications affected GRM1, GRM5, GRM7 and GRM8. The study concluded that "CNVs affecting metabotropic glutamate receptor genes were enriched across all cohorts (P = 2.1 × 10−9)", "over 200 genes interacting with glutamate receptors […] were collectively affected by CNVs", "major hubs of the (affected genes') network include TNIK50, GNAQ51, and CALM", and "the fact that children with ADHD are more likely to have alterations in these genes reinforces previous evidence that the GRM pathway is important in ADHD".[48]
A SciBX article in January 2012 commented that "UPenn and MIT teams have independently converged on mGluRs as players in ADHD and autism. The findings suggest agonizing mGluRs in patients with ADHD."[54]
Autism
The etiology of autism may include excessive glutamatergic mechanisms. In small studies, memantine has been shown to significantly improve language function and social behavior in children with autism.[55][56] Research is underway on the effects of memantine in adults with autism spectrum disorders.[57]
A link between glutamate receptors and autism was also identified via the structural protein ProSAP1 SHANK2 and potentially ProSAP2 SHANK3. The study authors concluded that the study "illustrates the significant role glutamatergic systems play in autism" and "By comparing the data on ProSAP1/Shank2−/− mutants with ProSAP2/Shank3αβ−/− mice, we show that different abnormalities in synaptic glutamate receptor expression can cause alterations in social interactions and communication. Accordingly, we propose that appropriate therapies for autism spectrum disorders are to be carefully matched to the underlying synaptopathic phenotype."[49]
Diabetes
Diabetes is a peculiar case because it is influenced by glutamate receptors present outside of the central nervous system, and it also influences glutamate receptors in the central nervous system.
Diabetes mellitus, an endocrine disorder, induces cognitive impairment and defects of long-term potential in the hippocampus, interfering with synaptic plasticity. Defects of long-term potential in the hippocampus are due to abnormal glutamate receptors, to be specific the malfunctioning NMDA glutamate receptors during early stages of the disease.[58]
Research is being done to address the possibility of using hyperglycemia and insulin to regulate these receptors and restore cognitive functions. Pancreatic islets regulating insulin and glucagon levels also express glutamate receptors.[28] Treating diabetes via glutamate receptor antagonists is possible, but not much research has been done. The difficulty of modifying peripheral GluR without having detrimental effects on the central nervous system, which is saturated with GluR, may be the cause of this.
Huntington's disease
In 2004, a specific genotype of human GluR6 was discovered to have a slight influence on the age of onset of Huntington's disease.[59]
In addition to similar mechanisms causing Parkinson's disease with respect to NMDA or AMPA receptors, Huntington's disease was also proposed to exhibit metabolic and mitochondrial deficiency, which exposes striatal neurons to the over activation of NMDA receptors.[38] Using folic acid has been proposed as a possible treatment for Huntington's due to the inhibition it exhibits on homocysteine, which increases vulnerability of nerve cells to glutamate.[60] This could decrease the effect glutamate has on glutamate receptors and reduce cell response to a safer level, not reaching excitotoxicity.
Ischemia
During ischemia, the brain has been observed to have an unnaturally high concentration of extracellular glutamate.[61] This is linked to an inadequate supply of ATP, which drives the glutamate transport levels that keep the concentrations of glutamate in balance.[62] This usually leads to an excessive activation of glutamate receptors, which may lead to neuronal injury. After this overexposure, the postsynaptic terminals tend to keep glutamate around for long periods of time, which results in a difficulty in depolarization.[62] Antagonists for NMDA and AMPA receptors seem to have a large benefit, with more aid the sooner it is administered after onset of the neural ischemia.[63]
Multiple sclerosis
Inducing experimental autoimmune encephalomyelitis in animals as a model for multiple sclerosis(MS) has targeted some glutamate receptors as a pathway for potential therapeutic applications.[64] This research has found that a group of drugs interact with the NMDA, AMPA, and kainate glutamate receptor to control neurovascular permeability, inflammatory mediator synthesis, and resident glial cell functions including CNS myelination. Oligodendrocytes in the CNS myelinate axons; the myelination dysfunction in MS is partly due to the excitotoxicity of those cells. By regulating the drugs which interact with those glutamate receptors, regulating glutamate binding may be possible, and thereby reduce the levels of Ca2+ influx. The experiments showed improved oligodendrocyte survival, and remyelination increased. Furthermore, CNS inflammation, apoptosis, and axonal damage were reduced.[64]
Parkinson's disease (Parkinsonism)
Late onset neurological disorders, such as Parkinson's disease, may be partially due to glutamate binding NMDA and AMPA glutamate receptors.[38] In vitro spinal cord cultures with glutamate transport inhibitors led to degeneration of motor neurons, which was counteracted by some AMPA receptor antagonists such as GYKI 52466.[38] Research also suggests that the metabotropic glutamate receptor mGlu4 is directly involved in movement disorders associated with the basal ganglia through selectively modulating glutamate in the striatum.[65]
Rasmussen's encephalitis
In 1994, GluR3 was shown to act as an autoantigen in Rasmussen's encephalitis, leading to speculation that autoimmune activity might underlie the condition.[34]
Schizophrenia
In schizophrenia, the expression of the mRNA for the NR2A subunit of the NMDA glutamate receptor was found to be decreased in a subset of inhibitory interneurons in the cerebral cortex.[66] This is suggested by upregulation of GABA, an inhibitory neurotransmitter. In schizophrenia, the expression of the NR2A subunit of NDMA receptors in mRNA was experimentally undetectable in 49-73% in GABA neurons that usually express it. These are mainly in GABA cells expressing the calcium-buffering protein parvalbumin (PV), which exhibits fast-spiking firing properties and target the perisomatic (basket cells) and axo-axonic (chandelier cells) compartments of pyramidal neurons.[66] The study found the density of NR2A mRNA-expressing PV neurons was decreased by as much as 50% in subjects with schizophrenia.
In addition, density of immunohistochemically labeled glutamatergic terminals with an antibody against the vesicular glutamate transporter vGluT1 also exhibited a reduction that paralleled the reduction in the NR2A-expressing PV neurons. Together, these observations suggest glutamatergic innervation of PV-containing inhibitory neurons appears to be deficient in schizophrenia.[66] Expression of NR2A mRNA has also been found to be altered in the inhibitory neurons that contain another calcium buffer, calbindin, targeting the dendrites of pyramidal neurons,[67] and the expression of the mRNA for the GluR5 kainate receptor in GABA neurons has also been found to be changed in people with schizophrenia.[68]
Current research is targeting glutamate receptor antagonists as potential treatments for schizophrenia. Memantine, a weak, nonselective NMDA receptor antagonist, was used as an add-on to clozapine therapy in a clinical trial. Refractory schizophrenia patients showed associated improvements in both negative and positive symptoms, underscoring the potential uses of GluR antagonists as antipsychotics.[69] Furthermore, administration of noncompetitive NMDA receptor antagonists have been tested on rat models. Scientists have proposed that specific antagonists can act on GABAergic interneurons, enhancing cortical inhibition and preventing excessive glutamatergic transmission associated with schizophrenia. These and other atypical antipsychotic drugs can be used together to inhibit excessive excitability in pyramidal cells, decreasing the symptoms of schizophrenia.[70]
Seizures
Glutamate receptors have been discovered to have a role in the onset of epilepsy. NMDA and metabotropic types have been found to induce epileptic convulsions. Using rodent models, labs have found that the introduction of antagonists to these glutamate receptors helps counteract the epileptic symptoms.[71] Since glutamate is a ligand for ligand-gated ion channels, the binding of this neurotransmitter will open gates and increase sodium and calcium conductance. These ions play an integral part in the causes of seizures. Group 1 metabotropic glutamate receptors (mGlu1 and mGlu5) are the primary cause of seizing, so applying an antagonist to these receptors helps in preventing convulsions.[72]
Other diseases suspected of glutamate receptor link
Neurodegenerative diseases with a suspected excitotoxicity link
Neurodegenerative diseases suspected to have a link mediated (at least in part) through stimulation of glutamate receptors:[36][73]
- AIDS dementia complex
- Alzheimer's disease
- Amyotrophic lateral sclerosis
- Combined systems disease (vitamin B12 deficiency)
- Depression/anxiety
- Drug addiction, tolerance, and dependency
- Essential tremor
- Glaucoma
- Hepatic encephalopathy
- Hydroxybutyric aminoaciduria
- Hyperhomocysteinemia and homocysteinuria
- Hyperprolinemia
- Lead encephalopathy
- Leber's disease
- MELAS syndrome
- MERRF
- Mitochondrial abnormalities (and other inherited or acquired biochemical disorders)
- Neuropathic pain syndromes (e.g. causalgia or painful peripheral neuropathies)
- Nonketotic hyperglycinemia
- Olivopontocerebellar atrophy (some recessive forms)
- Rett syndrome
- Sulfite oxidase deficiency
- Wernicke's encephalopathy
See also
References
- Brassai A, Suvanjeiev RG, Bán EG, Lakatos M (March 2015). "Role of synaptic and nonsynaptic glutamate receptors in ischaemia induced neurotoxicity". Review. Brain Research Bulletin. 112 (March 2015): 1–6. doi:10.1016/j.brainresbull.2014.12.007. PMID 25540918. S2CID 11292811.
- Petroff OA (December 2002). "GABA and glutamate in the human brain". Neuroscientist. 8 (6): 562–573. doi:10.1177/1073858402238515. PMID 12467378. S2CID 84891972. Glutamate is the main excitatory and GABA the main inhibitory neurotransmitter in the mammalian cortex
- "Glutamate Receptors - Structures and Functions". Centre of Synaptic Plasticity. University of Bristol. 2007-01-04. Archived from the original on 2007-09-15. Retrieved 2009-12-07.
- Watanabe M, Maemura K, Kanbara K, Tamayama T, Hayasaki H (2002). "GABA and GABA receptors in the central nervous system and other organs". A Survey of Cell Biology. pp. 1–47. doi:10.1016/S0074-7696(02)13011-7. ISBN 978-0-12-364617-0. PMID 11837891.
{{cite book}}:|journal=ignored (help) - Reiner A, Levitz J (June 2018). "Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert". Neuron. 98 (6): 1080–1098. doi:10.1016/j.neuron.2018.05.018. PMC 6484838. PMID 29953871.
- Debanne D, Daoudal G, Sourdet V, Russier M (2003). "Brain plasticity and ion channels". Journal of Physiology, Paris. 97 (4–6): 403–14. doi:10.1016/j.jphysparis.2004.01.004. PMID 15242652. S2CID 19116187.
- Maren S, Tocco G, Standley S, Baudry M, Thompson RF (October 1993). "Postsynaptic factors in the expression of long-term potentiation (LTP): increased glutamate receptor binding following LTP induction in vivo". Proceedings of the National Academy of Sciences of the United States of America. 90 (20): 9654–8. Bibcode:1993PNAS...90.9654M. doi:10.1073/pnas.90.20.9654. PMC 47628. PMID 8415757.
- Pérez-Otaño I, Ehlers MD (May 2005). "Homeostatic plasticity and NMDA receptor trafficking". Trends in Neurosciences. 28 (5): 229–38. doi:10.1016/j.tins.2005.03.004. PMID 15866197. S2CID 22901201.
- Asztély F, Gustafsson B (February 1996). "Ionotropic glutamate receptors. Their possible role in the expression of hippocampal synaptic plasticity". Molecular Neurobiology. 12 (1): 1–11. doi:10.1007/BF02740744. PMID 8732537. S2CID 42232786.
- Weiler IJ, Greenough WT (August 1993). "Metabotropic glutamate receptors trigger postsynaptic protein synthesis". Proceedings of the National Academy of Sciences of the United States of America. 90 (15): 7168–71. Bibcode:1993PNAS...90.7168W. doi:10.1073/pnas.90.15.7168. PMC 47097. PMID 8102206.
- Teichberg VI (December 1991). "Glial glutamate receptors: likely actors in brain signaling". FASEB Journal. 5 (15): 3086–91. doi:10.1096/fasebj.5.15.1660422. PMID 1660422. S2CID 15094280.
- Steinhäuser C, Gallo V (August 1996). "News on glutamate receptors in glial cells". Trends in Neurosciences. 19 (8): 339–45. doi:10.1016/0166-2236(96)10043-6. PMID 8843603. S2CID 31596399.
- Kandel ER (2013). Principles of neural science. McGraw Hill Professional. ISBN 9780071390118. OCLC 970875967.
- Suchanek B, Seeburg PH, Sprengel R (January 1995). "Gene structure of the murine N-methyl D-aspartate receptor subunit NR2C". J. Biol. Chem. 270 (1): 41–4. doi:10.1074/jbc.270.1.41. PMID 7814402. S2CID 6758394.
- Goto H, Watanabe K, Araragi N, Kageyama R, Tanaka K, Kuroki Y, Toyoda A, Hattori M, Sakaki Y, Fujiyama A, Fukumaki Y, Shibata H (2009). "The identification and functional implications of human-specific "fixed" amino acid substitutions in the glutamate receptor family". BMC Evol. Biol. 9: 224. doi:10.1186/1471-2148-9-224. PMC 2753569. PMID 19737383.
- Dingledine R, Borges K, Bowie D, Traynelis SF (March 1999). "The glutamate receptor ion channels". Pharmacol. Rev. 51 (1): 7–61. PMID 10049997.
- Andersson O, Stenqvist A, Attersand A, von Euler G (December 2001). "Nucleotide sequence, genomic organization, and chromosomal localization of genes encoding the human NMDA receptor subunits NR3A and NR3B". Genomics. 78 (3): 178–84. doi:10.1006/geno.2001.6666. PMID 11735224.
- Conn PJ, et al. (2005). "Metabotropic glutamate receptors in the basal ganglia motor circuit". Nature Reviews Neuroscience. 6 (10): 787–98. doi:10.1038/nrn1763. PMID 16276355. S2CID 9473598.
- Johnson JW, Ascher P (May 1990). "Voltage-dependent block by intracellular Mg2+ of N-methyl-D-aspartate-activated channels". Biophys. J. 57 (5): 1085–90. Bibcode:1990BpJ....57.1085J. doi:10.1016/S0006-3495(90)82626-6. PMC 1280813. PMID 1692749.
- Crowder JM, Croucher MJ, Bradford HF, Collins JF (June 1987). "Excitatory amino acid receptors and depolarization-induced Ca2+ influx into hippocampal slices". J. Neurochem. 48 (6): 1917–24. doi:10.1111/j.1471-4159.1987.tb05756.x. PMID 2437250. S2CID 12586076.
- Gover TD, Moreira TH, Weinreich D (2009). "Role of Calcium in Regulating Primary Sensory Neuronal Excitability". Sensory Nerves. Handbook of Experimental Pharmacology. Vol. 194. pp. 563–87. doi:10.1007/978-3-540-79090-7_16. ISBN 978-3-540-79089-1. PMID 19655118.
- Barbado M, Fablet K, Ronjat M, De Waard M (June 2009). "Gene regulation by voltage-dependent calcium channels" (PDF). Biochim. Biophys. Acta. 1793 (6): 1096–104. doi:10.1016/j.bbamcr.2009.02.004. PMID 19250948. S2CID 2807929.
- Muto T, Tsuchiya D, Morikawa K, Jingami H (March 2007). "Structures of the extracellular regions of the group II/III metabotropic glutamate receptors". Proc. Natl. Acad. Sci. U.S.A. 104 (10): 3759–64. Bibcode:2007PNAS..104.3759M. doi:10.1073/pnas.0611577104. PMC 1820657. PMID 17360426.
- Pin JP, Acher F (June 2002). "The metabotropic glutamate receptors: structure, activation mechanism and pharmacology". Curr Drug Targets CNS Neurol Disord. 1 (3): 297–317. doi:10.2174/1568007023339328. PMID 12769621.
- Platt SR (2007). "The role of glutamate in central nervous system health and disease--a review". Vet. J. 173 (2): 278–86. doi:10.1016/j.tvjl.2005.11.007. PMID 16376594.
- Kinnamon SC, Vandenbeuch A (July 2009). "Receptors and transduction of umami taste stimuli". Ann. N. Y. Acad. Sci. 1170 (1): 55–9. Bibcode:2009NYASA1170...55K. doi:10.1111/j.1749-6632.2009.04106.x. PMID 19686108. S2CID 20962248.
- Gill SS, Pulido OM, Mueller RW, McGuire PF (July 1998). "Molecular and immunochemical characterization of the ionotropic glutamate receptors in the rat heart". Brain Res. Bull. 46 (5): 429–34. doi:10.1016/S0361-9230(98)00012-4. PMID 9739005. S2CID 36044371.
- Weaver CD, Yao TL, Powers AC, Verdoorn TA (May 1996). "Differential expression of glutamate receptor subtypes in rat pancreatic islets". J. Biol. Chem. 271 (22): 12977–84. doi:10.1074/jbc.271.22.12977. PMID 8662728. S2CID 36816887.
- Bertrand G, Gross R, Puech R, Loubatières-Mariani MM, Bockaert J (June 1993). "Glutamate stimulates glucagon secretion via an excitatory amino acid receptor of the AMPA subtype in rat pancreas". Eur. J. Pharmacol. 237 (1): 45–50. doi:10.1016/0014-2999(93)90091-U. PMID 7689469.
- Weaver CD, Gundersen V, Verdoorn TA (January 1998). "A high affinity glutamate/aspartate transport system in pancreatic islets of Langerhans modulates glucose-stimulated insulin secretion". J. Biol. Chem. 273 (3): 1647–53. doi:10.1074/jbc.273.3.1647. PMID 9430708. S2CID 27129532.
- Carlton SM, Hargett GL, Coggeshall RE (September 1995). "Localization and activation of glutamate receptors in unmyelinated axons of rat glabrous skin". Neurosci. Lett. 197 (1): 25–8. doi:10.1016/0304-3940(95)11889-5. PMID 8545047. S2CID 44486341.
- Carlson NG, Gahring LC, Twyman RE, Rogers SW (April 1997). "Identification of amino acids in the glutamate receptor, GluR3, important for antibody-binding and receptor-specific activation". J. Biol. Chem. 272 (17): 11295–301. doi:10.1074/jbc.272.17.11295. PMID 9111034. S2CID 7239019.
- Gahring LC, Rogers SW, Twyman RE (February 1997). "Autoantibodies to glutamate receptor subunit GluR2 in nonfamilial olivopontocerebellar degeneration". Neurology. 48 (2): 494–500. doi:10.1212/wnl.48.2.494. PMID 9040745. S2CID 20274921.
- Autoantibodies to glutamate receptor GluR3 in Rasmussen's encephalitis - 1994
- He XP, Patel M, Whitney KD, Janumpalli S, Tenner A, McNamara JO (January 1998). "Glutamate receptor GluR3 antibodies and death of cortical cells". Neuron. 20 (1): 153–63. doi:10.1016/S0896-6273(00)80443-2. PMID 9459451. S2CID 1742781.
- Dubinsky JM (February 1993). "Intracellular calcium levels during the period of delayed excitotoxicity". J. Neurosci. 13 (2): 623–31. doi:10.1523/JNEUROSCI.13-02-00623.1993. PMC 6576661. PMID 8093901.
- Manev H, Favaron M, Guidotti A, Costa E (July 1989). "Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death". Mol. Pharmacol. 36 (1): 106–12. PMID 2568579.
- Meldrum B (1993). "Amino acids as dietary excitotoxins: a contribution to understanding neurodegenerative disorders". Brain Res. Brain Res. Rev. 18 (3): 293–314. doi:10.1016/0165-0173(93)90014-Q. PMID 8401596. S2CID 11003515.
- Aoyama K, Watabe M, Nakaki T (November 2008). "Regulation of neuronal glutathione synthesis". J. Pharmacol. Sci. 108 (3): 227–38. doi:10.1254/jphs.08R01CR. PMID 19008644.
- Markowitz AJ, White MG, Kolson DL, Jordan-Sciutto KL (July 2007). "Cellular interplay between neurons and glia: toward a comprehensive mechanism for excitotoxic neuronal loss in neurodegeneration". Cellscience. 4 (1): 111–146. PMC 2613343. PMID 19122795.
- Nicholls DG (December 2009). "Spare respiratory capacity, oxidative stress and excitotoxicity". Biochem. Soc. Trans. 37 (Pt 6): 1385–8. doi:10.1042/BST0371385. PMID 19909281. S2CID 14826555.
- Hirsch JA, Gibson GE (August 1984). "Selective alteration of neurotransmitter release by low oxygen in vitro". Neurochem. Res. 9 (8): 1039–49. doi:10.1007/BF00964800. PMID 6149480. S2CID 22594469.
- Obrenovitch TP, Richards DA (1995). "Extracellular neurotransmitter changes in cerebral ischaemia". Cerebrovasc Brain Metab Rev. 7 (1): 1–54. PMID 7742171.
- Beal MF (December 1992). "Mechanisms of excitotoxicity in neurologic diseases". FASEB J. 6 (15): 3338–44. doi:10.1096/fasebj.6.15.1464368. PMID 1464368. S2CID 22958157.
- Nechifor M (March 2008). "Magnesium in drug dependences". Magnesium Research. 21 (1): 5–15. doi:10.1684/mrh.2008.0124 (inactive 1 August 2023). PMID 18557129.
{{cite journal}}: CS1 maint: DOI inactive as of August 2023 (link) - "Glutamate Receptor Summary Report". CureHunter. Retrieved 2009-12-07.
- Vadasz C, Saito M, Gyetvai BM, Oros M, Szakall I, Kovacs KM, Prasad VV, Toth R (2007). "Glutamate receptor metabotropic 7 is cis-regulated in the mouse brain and modulates alcohol drinking". Genomics. 90 (6): 690–702. doi:10.1016/j.ygeno.2007.08.006. PMID 17936574.
- Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D, Sleiman PM, Zhang H, Kim CE, Robison R, et al. (January 2012). "Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder". Nat. Genet. 44 (1): 78–84. doi:10.1038/ng.1013. PMC 4310555. PMID 22138692.
- "Glutamate Receptor Gene Changes Identified in ADHD". Genetic Engineering & Biotechnology News. December 5, 2011.
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, et al. (June 2012). "Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2". Nature. 486 (7402): 256–60. Bibcode:2012Natur.486..256S. doi:10.1038/nature11015. PMID 22699619. S2CID 4412353.
- "Of Mice and Men: Characterization of a new autism gene". AlphaGalileo Foundation (Press release). 3 May 2012. Archived from the original on 2012-05-17.
- Plumb B. "Glutamate - A Pain Transmitter Your Subconscious May Be Able To Limit". EzineArticles.com.
{{cite web}}: Missing or empty|url=(help) - Dorval KM, Wigg KG, Crosbie J, Tannock R, Kennedy JL, Ickowicz A, Pathare T, Malone M, Schachar R, Barr CL (July 2007). "Association of the glutamate receptor subunit gene GRIN2B with attention-deficit/hyperactivity disorder". Genes Brain Behav. 6 (5): 444–52. doi:10.1111/j.1601-183X.2006.00273.x. PMC 4833492. PMID 17010153.
- Gainetdinov RR, Mohn AR, Bohn LM, Caron MG (September 2001). "Glutamatergic modulation of hyperactivity in mice lacking the dopamine transporter". Proc. Natl. Acad. Sci. U.S.A. 98 (20): 11047–54. Bibcode:2001PNAS...9811047G. doi:10.1073/pnas.191353298. PMC 58681. PMID 11572967.
- Turic D, Langley K, Williams H, Norton N, Williams NM, Moskvina V, Van den Bree MB, Owen MJ, Thapar A, O'Donovan MC (June 2005). "A family based study implicates solute carrier family 1-member 3 (SLC1A3) gene in attention-deficit/hyperactivity disorder". Biol. Psychiatry. 57 (11): 1461–6. doi:10.1016/j.biopsych.2005.03.025. PMID 15950021. S2CID 26867098.
- Osherovich L (2012). "Fine-tuning mGluRs". Science-Business EXchange. 5 (1): 3. doi:10.1038/scibx.2012.3.
- Chez MG, Burton Q, Dowling T, Chang M, Khanna P, Kramer C (May 2007). "Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability". Journal of Child Neurology. 22 (5): 574–9. doi:10.1177/0883073807302611. PMID 17690064. S2CID 41428656.
- Erickson CA, Posey DJ, Stigler KA, Mullett J, Katschke AR, McDougle CJ (March 2007). "A retrospective study of memantine in children and adolescents with pervasive developmental disorders". Psychopharmacology. 191 (1): 141–7. doi:10.1007/s00213-006-0518-9. PMID 17016714. S2CID 13128356.
- Clinical trial number NCT01333865 for "A Study of Memantine Hydrochloride (Namenda®) for Cognitive and Behavioral Impairment in Adults With Autism Spectrum Disorders" at ClinicalTrials.gov
- Trudeau F, Gagnon S, Massicotte G (April 2004). "Hippocampal synaptic plasticity and glutamate receptor regulation: influences of diabetes mellitus". Eur. J. Pharmacol. 490 (1–3): 177–86. doi:10.1016/j.ejphar.2004.02.055. PMID 15094084.
- Diguet E, Fernagut PO, Normand E, Centelles L, Mulle C, Tison F (April 2004). "Experimental basis for the putative role of GluR6/kainate glutamate receptor subunit in Huntington's disease natural history". Neurobiol. Dis. 15 (3): 667–75. doi:10.1016/j.nbd.2003.12.010. PMID 15056475. S2CID 41752581.
- Wu J, Tang T, Bezprozvanny I (October 2006). "Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington's disease". Neurosci. Lett. 407 (3): 219–23. doi:10.1016/j.neulet.2006.08.036. PMID 16959411. S2CID 18046017.
- "Test reveals effectiveness of potential Huntington's disease drugs". Innovations Report. 2006-10-31.
- Nishizawa Y. (2001). "Glutamate release and neuronal damage in ischemia". Life Sci. 69 (4): 369–81. doi:10.1016/S0024-3205(01)01142-0. PMID 11459428.
- Ottersen OP. "Sources of Glutamate in Ischemia". Neurodegeneration Research Group. University of Oslo. Archived from the original on 1998-12-02. Retrieved 2009-12-07.
- Meldrum BS (2000). "Glutamate as a neurotransmitter in the brain: review of physiology and pathology". J. Nutr. 130 (4S Suppl): 1007S–15S. doi:10.1093/jn/130.4.1007s. PMID 10736372.
- Bolton C, Paul C (2006). "Glutamate receptors in neuroinflammatory demyelinating disease". Mediators of Inflammation. 2006 (2): 93684. doi:10.1155/MI/2006/93684. PMC 1592583. PMID 16883070.
- Cuomo D, et al. (2009). "Metabotropic glutamate receptor subtype 4 selectively modulates both glutamate and GABA transmission in the striatum: implications for Parkinson's disease treatment". J Neurochem. 109 (4): 1096–105. doi:10.1111/j.1471-4159.2009.06036.x. PMID 19519781. S2CID 23414211.
- Bitanihirwe BK, Lim MP, Kelley JF, Kaneko T, Woo TU (November 2009). "Glutamatergic deficits and parvalbumin-containing inhibitory neurons in the prefrontal cortex in schizophrenia". BMC Psychiatry. 9: 71. doi:10.1186/1471-244X-9-71. PMC 2784456. PMID 19917116.
- Woo TU, Shrestha K, Lamb D, Minns MM, Benes FM (April 2008). "N-Methyl-D-Aspartate Receptor and Calbindin-Containing Neurons in the Anterior Cingulate Cortex in Schizophrenia and Bipolar Disorder". Biol Psychiatry. 64 (9): 803–809. doi:10.1016/j.biopsych.2008.04.034. PMC 3877780. PMID 18585682.
- Woo TU, Shrestha K, Armstrong C, Minns MM, Walsh JP, Benes FM (August 2007). "Differential alterations of kainate receptor subunits in inhibitory interneurons in the anterior cingulate cortex in schizophrenia and bipolar disorder". Schizophrenia Research. 96 (1–3): 46–61. doi:10.1016/j.schres.2007.06.023. PMC 2712609. PMID 17698324.
- de Lucena D, Fernandes BS, Berk M, et al. (October 2009). "Improvement of negative and positive symptoms in treatment-refractory schizophrenia: a double-blind, randomized, placebo-controlled trial with memantine as add-on therapy to clozapine". J Clin Psychiatry. 70 (10): 1416–23. doi:10.4088/JCP.08m04935gry. PMID 19906345. S2CID 26374475.
- López-Gil X, Artigas F, Adell A (November 2009). "Unraveling Monoamine Receptors Involved in the Action of Typical and Atypical Antipsychotics on Glutamatergic and Serotonergic Transmission in Prefrontal Cortex". Curr. Pharm. Des. 16 (5): 502–15. doi:10.2174/138161210790361416. hdl:10261/77721. PMID 19909228.
- Chapman AG (April 2000). "Glutamate and epilepsy". J. Nutr. 130 (4S Suppl): 1043S–5S. doi:10.1093/jn/130.4.1043S. PMID 10736378.
- Moldrich RX, Chapman AG, De Sarro G, Meldrum BS (August 2003). "Glutamate metabotropic receptors as targets for drug therapy in epilepsy". Eur. J. Pharmacol. 476 (1–2): 3–16. doi:10.1016/S0014-2999(03)02149-6. PMID 12969743.
- Gillessen T, Budd SL, Lipton SA (2003). "Excitatory Amino Acid Neurotoxicity". Molecular and Cellular Biology of Neuroprotection in the CNS. Advances in Experimental Medicine and Biology. Vol. 513. pp. 3–40. doi:10.1007/978-1-4615-0123-7_1. ISBN 978-1-4613-4934-1. PMID 12575816.
External links
- Glutamate+Receptors at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- "Metabotropic Glutamate Receptors". IUPHAR Database of Receptors and Ion Channels. International Union of Basic and Clinical Pharmacology.