Doxepin
Doxepin is a medication belonging to the tricyclic antidepressant (TCA)[10] class of drugs used to treat major depressive disorder, anxiety disorders, chronic hives, and insomnia.[10][11] For hives it is a less preferred alternative to antihistamines.[10] It has a mild to moderate benefit for sleeping problems.[12] It is used as a cream for itchiness due to atopic dermatitis or lichen simplex chronicus.[13]
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Sinequan, Quitaxon, Adapin, Triadapin, Aponal, others[1] |
| Other names | NSC-108160[2] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682390 |
| License data | |
| Pregnancy category |
|
| Routes of administration | By mouth, topical, intravenous, intramuscular injection[3] |
| Drug class | Tricyclic antidepressant (TCA) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 13–45% (mean 29%)[5][6] |
| Protein binding | 76%[7] |
| Metabolism | Hepatic (CYP2D6, CYP2C19)[8][5] |
| Metabolites | Nordoxepin, glucuronide conjugates[8] |
| Elimination half-life | Doxepin: 8–24 hours (mean 17 hours)[7] Nordoxepin: 28–31 hours[7][9] |
| Excretion | Urine: ~50%[8][5] Feces: minor[5] |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
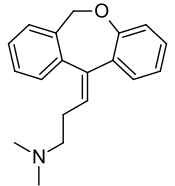
| Formula | C19H21NO |
| Molar mass | 279.383 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Common side effects include sleepiness, dry mouth, constipation, nausea, and blurry vision.[10] Serious side effects may include increased risk of suicide in those under the age of 25, mania, and urinary retention.[10] A withdrawal syndrome may occur if the dose is rapidly decreased.[10] Use during pregnancy and breastfeeding is not generally recommended.[14][15] Although how it works for treating depression remains an area of active inquiry, it may involve increasing the levels of norepinephrine, along with blocking histamine, acetylcholine, and serotonin.[10]
Doxepin was approved for medical use in the United States in 1969.[10] It is available as a generic medication.[14][16][17] In 2020, it was the 252nd most commonly prescribed medication in the United States, with more than 1 million prescriptions.[18][19]
Medical uses
Doxepin is used as a pill to treat major depressive disorder, anxiety disorders, and chronic hives, and for short-term help with trouble remaining asleep after going to bed (a form of insomnia).[10][7][11] As a cream it is used for short-term treatment of itchiness caused by atopic dermatitis or lichen simplex chronicus.[13]
Insomnia
Doxepin is used in the treatment of insomnia.[11] In 2016, the American College of Physicians advised that insomnia be treated first by treating comorbid conditions, then with cognitive behavioral therapy and behavioral changes, and then with drugs; doxepin was among those recommended for short-term help maintaining sleep, on the basis of weak evidence.[20][21] The 2017 American Academy of Sleep Medicine recommendations focused on treatment with drugs were similar.[20] A 2015 Agency for Healthcare Research and Quality review of treatments for insomnia had similar findings.[22]
A major systematic review and network meta-analysis of medications for the treatment of insomnia published in 2022 found that doxepin had an effect size (standardized mean difference (SMD)) against placebo for treatment of insomnia at 4 weeks of 0.30 (95% CITooltip confidence interval –0.05 to 0.64).[23] The certainty of evidence was rated as very low, and no data were available for longer-term treatment (3 months).[23] For comparison, the other sedating antihistamines assessed, trimipramine and doxylamine, had effect sizes (SMD) at 4 weeks of 0.55 (95% CI –0.11 to 1.21) (very low certainty evidence) and 0.47 (95% CI 0.06 to 0.89) (moderate certainty evidence), respectively.[23] Benzodiazepines and Z-drugs generally showed larger effect sizes (e.g., SMDs of 0.45 to 0.83) than doxepin, whereas the effect sizes of orexin receptor antagonists, such as suvorexant, were more similar (SMDs of 0.23 to 0.44).[23]
Doses of doxepin used for sleep normally range from 3 to 6 mg, but high doses of up to 25 to 50 mg may be used as well.[24][25]
Contraindications
Known contraindications include:[28]
- Hypersensitivities to doxepin, other TCAs, or any of the excipients inside the product used
- Glaucoma
- A predisposition to developing urinary retention such as in benign prostatic hyperplasia
- Use of monoamine oxidase inhibitors in last 14 days[29]
Pregnancy and lactation
Its use in pregnant and lactating women is advised against, although the available evidence suggests it is unlikely to cause negative effects on fetal development.[7] The lack of evidence from human studies, however, means it is currently impossible to rule out any risk to the fetus and it is known to cross the placenta.[7] Doxepin is secreted in breast milk[3] and neonatal cases of respiratory depression in association with maternal doxepin use have been reported.[30]
Side effects
Doxepin's side effects profile may differ from the list below in some countries where it is licensed to be used in much smaller doses (viz., 3 mg and 6 mg).
- Central nervous system: fatigue, dizziness, drowsiness, lightheadedness, confusion, nightmares, agitation, increased anxiety, difficulty sleeping, seizures (infrequently), temporary confusion (delirium), rarely induction of hypomania and schizophrenia (stop medication immediately), extrapyramidal side effects (rarely), abuse in patients with polytoxicomania (rarely), ringing in the ears (tinnitus)
- Anticholinergic: dry mouth, constipation, even ileus (rarely), difficulties in urinating, sweating, precipitation of glaucoma
- Antiadrenergic: Low blood pressure, (if patient arises too fast from the lying/sitting position to standing—known as orthostatic hypotension), abnormal heart rhythms (e.g., sinus tachycardia, bradycardia, and atrioventricular block)
- Allergic/toxic: skin rash, photosensitivity, liver damage of the cholestatic type (rarely), hepatitis (extremely rare), leuko- or thrombocytopenia (rarely), agranulocytosis (very rarely), hypoplastic anemia (rarely)
- Others: frequently increased appetite and weight gain, rarely nausea, rarely high blood pressure. May increase or decrease liver enzyme levels in the blood of some people.[31]
The side effects of low-dose doxepin for insomnia in long-term clinical trials (28 to 85 days) in adults and elderly people were as follows:[11]
| Side effect | Placebo (N=278) | Doxepin 3 mg (N=157) | Doxepin 6 mg (N=203) |
|---|---|---|---|
| Somnolence/sedation | 4% | 6% | 9% |
| Upper respiratory tract infection or nasopharyngitis | 2% | 4% | 2% |
| Gastroenteritis | 0% | 2% | 0% |
| Nausea | 1% | 2% | 2% |
| Hypertension | 0% | 3% | <1% |
| Note: Includes reactions that occurred at a rate of ≥ 2% in any doxepin-treated group and at a higher rate than placebo. | |||
Overdose
Like other TCAs, doxepin is highly toxic in cases of overdose.[32] Mild symptoms include drowsiness, stupor, blurred vision, and excessive dryness of mouth. More serious adverse effects include respiratory depression, hypotension, coma, convulsions, cardiac arrhythmia, and tachycardia. Urinary retention, decreased gastrointestinal motility (paralytic ileus), hyperthermia (or hypothermia), hypertension, dilated pupils, and hyperactive reflexes are other possible symptoms of doxepin overdose.[7] Management of overdose is mostly supportive and symptomatic, and can include the administration of a gastric lavage so as to reduce absorption of the doxepin.[7] Supportive measures to prevent respiratory aspiration is also advisable.[7] Antiarrhythmic agents may be an appropriate measure to treat cardiac arrhythmias resulting from doxepin overdose.[7] Slow intravenous administration of physostigmine may reverse some of the toxic effects of overdose such as anticholinergic effects.[7] Haemodialysis is not recommended due to the high degree of protein binding with doxepin.[7] ECG monitoring is recommended for several days after doxepin overdose due to the potential for cardiac conduction abnormalities.[7]
Interactions
Doxepin should not be used within 14 days of using a monoamine oxidase inhibitor (MAOI) such as phenelzine due to the potential for hypertensive crisis or serotonin syndrome to develop.[28] Its use in those taking potent CYP2D6 inhibitors such as fluoxetine, paroxetine, sertraline, duloxetine, bupropion, and quinidine is recommended against owing to the potential for its accumulation in the absence of full CYP2D6 catalytic activity.[28][33] Hepatic enzyme inducers such as carbamazepine, phenytoin, and barbiturates are advised against in patients receiving TCAs like doxepin owing to the potential for problematically rapid metabolism of doxepin to occur in these individuals.[28] Sympathomimetic agents may have their effects potentiated by TCAs like doxepin.[28] Doxepin also may potentiate the adverse effects of anticholinergic agents such as benztropine, atropine and hyoscine (scopolamine).[28] Tolazamide, when used in conjunction with doxepin has been associated with a case of severe hypoglycaemia in a type II diabetic individual.[28] Cimetidine may influence the absorption of doxepin.[28] Alcohol may potentiate some of the CNS depressant effects of doxepin.[28] Antihypertensive agents may have their effects mitigated by doxepin.[28] Cotreatment with CNS depressants such as the benzodiazepines can cause additive CNS depression.[7] Co-treatment with thyroid hormones may also increase the potential for adverse reactions.[7]
Pharmacology
Doxepin is a tricyclic antidepressant (TCA).[10] It acts as a serotonin–norepinephrine reuptake inhibitor (SNRI) (a reuptake inhibitor of serotonin and norepinephrine), with additional antiadrenergic, antihistamine, antiserotonergic, and anticholinergic activities.[34][35]
Pharmacodynamics
| Site | Ki (nM) | Species | Ref |
|---|---|---|---|
| SERTTooltip Serotonin transporter | 68–95 210 (IC50Tooltip Half-maximal inhibitory concentration) | Human | [36][34] [8] |
| NETTooltip Norepinephrine transporter | 30–58 13 (IC50) | Human | [36][34] [8] |
| DATTooltip Dopamine transporter | >10,000 4,600 (IC50) | Human | [36] [8] |
| 5-HT1A | 276 | Human | [37] |
| 5-HT2A | 11–27 | Human | [34][37] |
| 5-HT2B | ND | ND | ND |
| 5-HT2C | 200 8.8 | Human Rat | [34] [38] |
| 5-HT3 | ND | Human | [39] |
| 5-HT6 | 136 | Rat | [40] |
| 5-HT7 | ND | ND | ND |
| α1 | 24 | Human | [34] |
| α1B | 12 | Human | [34] |
| α2A | 1,100–1,270 | Human | [34][37] |
| α2B | 28 | Human | [34] |
| α2C | 96 | Human | [34] |
| D2 | 360 | Human | [37] |
| H1 | 0.09–1.23 | Human | [41][37][34] |
| H2 | 174 | Human | [41] |
| H3 | 39,800 | Human | [41][34] |
| H4 | 15,100 | Human | [41][42] |
| mAChTooltip Muscarinic acetylcholine receptor | 23–80 | Human | [37][43] |
| M1 | 18–38 | Human | [34][44] |
| M2 | 160–230 | Human | [34][44] |
| M3 | 25–52 | Human | [34][44] |
| M4 | 20–82 | Human | [34][44] |
| M5 | 5.6–75 | Human | [34][44] |
| hERGTooltip Human Ether-à-go-go-Related Gene | 6,500 (IC50Tooltip Half-maximal inhibitory concentration) | Human | [45] |
| Values are Ki, unless otherwise specified. The smaller the value, the more strongly the drug binds to the site. | |||
Doxepin is a reuptake inhibitor of serotonin and norepinephrine, or a serotonin–norepinephrine reuptake inhibitor (SNRI), and has additional antiadrenergic, antihistamine, antiserotonergic, and anticholinergic activities.[34][35] It is specifically an antagonist of the histamine H1 and H2 receptors, the serotonin 5-HT2A and 5-HT2C receptors, the α1-adrenergic receptor, and the muscarinic acetylcholine receptors (M1–M5).[35] Similarly to other tricyclic antidepressants, doxepin is often prescribed as an effective alternative to SSRI medications. Doxepin is also a potent blocker of voltage-gated sodium channels, and this action is thought to be involved in both its lethality in overdose[46] and its effectiveness as an analgesic (including in the treatment of neuropathic pain,[47] and as a local anesthetic).[48] The potencies of doxepin in terms of its receptor antagonism specifically are as follows:[48][49]
- Extremely strong: Histamine H1 receptor
- Strong: α1-adrenergic receptor, 5-HT2A and muscarinic acetylcholine receptors
- Moderate: 5-HT2C and 5-HT1A receptors
- Weak: α2-adrenergic and D2 receptors
Based on its IC50Tooltip half-maximal inhibitory concentration values for monoamine reuptake inhibition, doxepin is relatively selective for the inhibition of norepinephrine reuptake, with a much weaker effect on the serotonin transporter. Although there is a significant effect that takes place at one of the specific serotonergic binding sites, the 5-HT2A serotonin receptor subtype. There is negligible influence on dopamine reuptake.[36][34]
The major metabolite of doxepin, nordoxepin (desmethyldoxepin), is pharmacologically active similarly,[8] but relative to doxepin, is much more selective as a norepinephrine reuptake inhibitor.[50][51] In general, the demethylated variants of tertiary amine TCAs like nordoxepin are much more potent inhibitors of norepinephrine reuptake, less potent inhibitors of serotonin reuptake, and less potent in their antiadrenergic, antihistamine, and anticholinergic activities.[50][51][52]
Antidepressant doses of doxepin are defined as 25 to 300 mg/day, although are typically above 75 mg/day.[53][54] Antihistamine doses, including for dermatological uses and as a sedative/hypnotic for insomnia, are considered to be 3 to 25 mg,[55][54] although higher doses between 25 and 50 mg and in some cases even up to 150 mg have been used to treat insomnia.[56] At low doses, below 25 mg, doxepin is a pure antihistamine and has more of a sedative effect.[53] At antidepressant doses of above 75 mg, doxepin is more stimulating with antiadrenergic, antiserotonergic, and anticholinergic effects, and these activities contribute to its side effects.[55][53][54]
Doxepin is a mixture of (E) and (Z) stereoisomers with an approximate ratio of 85:15.[5] When doxepin was developed, no effort was made to separate or balance the mixture following its synthesis, resulting in the asymmetric ratio.[5] (Z)-Doxepin is more active as an inhibitor of serotonin and norepinephrine reuptake than (E)-doxepin.[5] The selectivity of doxepin for inhibition of norepinephrine reuptake over that of serotonin is likely due to the 85% presence of (E)-doxepin in the mixture.[5] Most other tertiary amine TCAs like amitriptyline and imipramine do not exhibit E-Z isomerism or such mixture asymmetry and are comparatively more balanced inhibitors of serotonin and norepinephrine reuptake.[5][36]
As a hypnotic
| Drug | H1 | mAChTooltip Muscarinic acetylcholine receptor | Ratio |
|---|---|---|---|
| Amitriptyline | 1.1 | 18 | 1:16 |
| Amoxapine | 25 | 1,000 | 1:40 |
| Clomipramine | 31 | 37 | 1:1.2 |
| Desipramine | 110 | 196 | 1:1.8 |
| Dosulepin[55] | 4.0 | 38 | 1:9.5 |
| Doxepin | 0.24 | 83 | 1:346 |
| Imipramine | 11 | 91 | 1:8.3 |
| Lofepramine[37] | 360 | 67 | 1:0.2 |
| Maprotiline | 2.0 | 560 | 1:280 |
| Mianserin | 0.40 | 820 | 1:2050 |
| Mirtazapine | 0.14 | 670 | 1:4786 |
| Nortriptyline | 10 | 149 | 1:15 |
| Protriptyline | 25 | 25 | 1:1 |
| Trimipramine | 0.27 | 58 | 1:215 |
| Values are Ki (nM). | |||
Doxepin is a highly potent antihistamine, with this being its strongest activity.[49][53][58][8] In fact, doxepin has been said to be the most or one of the most potent H1 receptor antagonists available, with one study finding an in vitro Ki of 0.17 nM.[37] It is the most potent and selective H1 receptor antagonist of the TCAs (although the tetracyclic antidepressant (TeCA) mirtazapine is slightly more potent),[55][59][60] and other sedating antihistamines, for instance the over-the-counter diphenhydramine (Ki = 16 nM) and doxylamine (Ki = 42 nM), show far lower affinities for this receptor in comparison.[8] The affinity of doxepin for the H1 receptor is far greater than its affinity for other sites,[8] and 10- to 100-fold higher doses are needed for antidepressant effects.[61][58] In accordance, although it is often described as a "dirty drug" due to its highly promiscuous binding profile,[58] doxepin acts as a highly selective antagonist of the H1 receptor at very low doses (less than 10 mg; typically 3 to 6 mg).[53][8][54] At these doses, it notably has no clinically relevant anticholinergic effects such as dry mouth or cognitive/memory impairment, unlike most other sedating antihistamines, and similarly has no effect on other receptors such as adrenergic and serotonin receptors.[53][8][54]
The H1 receptor antagonism of doxepin is responsible for its hypnotic effects and its effectiveness in the treatment of insomnia at low doses.[8][58] The incidence of side effects with doxepin and its safety at these doses was similar to that of placebo in clinical trials; the most frequent side effects were headache and somnolence/sedation, both with an incidence of less than 5%.[53][8] Other side effects sometimes associated with antihistamines, including daytime sedation, increased appetite, and weight gain, all were not observed.[58] Clinical evidence of H1 receptor antagonists and TCAs for the treatment insomnia shows mixed effectiveness and is limited in its quality due to weaknesses like small sample sizes and poor generalizability.[54][62] However, doxepin is a unique and notable exception; it has been well-studied in the treatment of insomnia and shows consistent benefits with excellent tolerability and safety.[54][62] Aside from diphenhydramine and doxylamine, which have historical approval as hypnotics, doxepin is the only H1 receptor antagonist that is specifically approved for the treatment of insomnia in the United States.[62][63]
The effect sizes of very low-dose doxepin in the treatment of insomnia range from small to medium.[54] These include subjective and objective measures of sleep maintenance, sleep duration, and sleep efficiency.[54] Conversely, very low-dose doxepin shows relatively weak effects on sleep initiation and does not significantly separate from placebo on this measure.[54] This is in contrast to benzodiazepines and nonbenzodiazepine (Z-drug) hypnotics, which are additionally effective in improving sleep onset latency.[54] However, it is also in contrast to higher doses of doxepin (50 to 300 mg/day), which have been found to significantly reduce latency to sleep onset.[54] A positive dose–response relationship on sleep measures was observed for doses of doxepin between 1 and 6 mg in clinical studies, whereas the incidence of adverse effects remained constant across this dose range in both young and older adults.[54] However, the incidence of adverse effects appeared to increase with longer treatment duration.[54] A dose of doxepin as low as 1 mg/day was found to significantly improve most of the assessed sleep measures, but unlike the 3 and 6 mg/day doses, was not able to improve wake time during sleep.[54] This, along with greater effect sizes with the higher doses, was likely the basis for the approval of the 3 and 6 mg doses of doxepin for insomnia and not the 1 mg dose.[54]
At very low doses, doxepin has not shown discontinuation or withdrawal effects nor rebound insomnia.[8] Sustained effectiveness without apparent tolerance was demonstrated in clinical studies of up to 12 weeks duration.[62] This appears to be in contrast to over-the-counter antihistamines like diphenhydramine and doxylamine and all other first-generation antihistamines, which are associated with rapid development of tolerance and dependence (by day 3 or 4 of continuous dosing) and loss of hypnotic effectiveness.[62] It is for this reason that, unlike doxepin, they are not recommended for the chronic management of insomnia and are advised for only short-term treatment (i.e., 1 week).[62] It is not entirely clear why doxepin and first-generation antihistamines are different in this regard, but it has been suggested that it may have to do with the lack of selectivity for the H1 receptor of the latter or may have to do with the use of optimal doses.[58] Unlike very-low-dose doxepin, most first-generation antihistamines also have marked anticholinergic activity as well as associated side effects such as dry mouth, constipation, urinary retention, and confusion.[62] This is particularly true in older people, and antihistamines with concomitant anticholinergic effects are not recommended in adults over the age of 65.[62] Anticholinergic activity notably may interfere with the sleep-promoting effects of H1 receptor blockade.[34]
Antagonism of the H1, 5-HT2A, 5-HT2C, and α1-adrenergic receptors is thought to have sleep-promoting effects and to be responsible for the sedative effects of TCAs including those of doxepin.[64][65][66] Although doxepin is selective for the H1 receptor at doses lower than 25 mg, blockade of serotonin and adrenergic receptors may also be involved in the hypnotic effects of doxepin at higher doses.[64] However, in contrast to very low doses of doxepin, rebound insomnia and daytime sedation are significantly more frequent than placebo with moderate doses (25 to 50 mg/day) of the drug.[54] In addition, one study found that although such doses of doxepin improved sleep measures initially, most of the benefits were lost with chronic treatment (by 4 weeks).[54] Due to limited data however, more research on potential tolerance and withdrawal effects of moderate doses of doxepin is needed.[54] At these doses of doxepin, dry mouth, an anticholinergic effect, was common (71%), and other side effects such as headache (25%), increased appetite (21%), and dizziness (21%) were also frequently observed, although these adverse effects were notably not significantly more frequent than with placebo in the study in question.[54] In any case, taken together, higher doses of doxepin than very low doses are associated with an increased rate of side effects as well as apparent loss of hypnotic effectiveness with chronic treatment.[58]
Doxepin at a dose of 25 mg/day for 3 weeks has been found to decrease cortisol levels by 16% in adults with chronic insomnia and to increase melatonin production by 26% in healthy volunteers.[8] In individuals with neuroendocrine dysregulation in the form of nocturnal melatonin deficiency presumably due to chronic insomnia, very-low-dose doxepin was found to restore melatonin levels to near-normal values after 3 weeks of treatment.[48] These findings suggest that normalization of the hypothalamic–pituitary–adrenal axis and the circadian sleep–wake cycle may be involved in the beneficial effects of doxepin on sleep and insomnia.[8][48]
CYP2D6 inhibition
Doxepin has been identified as an inhibitor of CYP2D6 in vivo in a study of human patients being treated with 75 to 250 mg/day for depression.[67] While it significantly altered metabolic ratios for sparteine and its metabolites, doxepin did not convert any of the patients to a different metabolizer phenotype (e.g., extensive to intermediate or poor).[67] Nonetheless, inhibition of CYP2D6 by doxepin could be of clinical importance.[67]
Pharmacokinetics
| Parameters | Doxepin | Nordoxepin |
|---|---|---|
| TmaxTooltip Time to peak concentrations (hours) | Mean: 2.9 Range: 2–4 | Mean: ND Range: 2–10 |
| CmaxTooltip Peak concentrations (ng/mL) | Mean: ND Range: 8.8–45.8 | Mean: 9.7 Range: 4.8–14.5 |
| VDTooltip Volume of distribution (L/kg) | 20 | ND |
| Protein bindingTooltip Plasma protein binding | 76% | ND |
| t1/2Tooltip Terminal half-life (hours) | Mean: 17 Range: 8–24 | Mean: 31 Range: ND |
| Metabolic enzymes | Major: CYP2D6, CYP2C19 Minor: CYP1A2, CYP2C9, CYP3A4 | |
| Metabolic pathways | N-Demethylation, N-oxidation, hydroxylation, glucuronidation | |
Absorption
Doxepin is well-absorbed from the gastrointestinal tract but between 55 and 87% undergoes first-pass metabolism in the liver,[8] resulting in a mean oral bioavailability of approximately 29%.[6] Following a single very low dose of 6 mg, peak plasma levels of doxepin are 0.854 ng/mL (3.06 nmol/L) at 3 hours without food and 0.951 ng/mL (3.40 nmol/L) at 6 hours with food.[8] Plasma concentrations of doxepin with antidepressant doses are far greater, ranging between 50 and 250 ng/mL (180 to 900 nmol/L).[68] Area-under-curve levels of the drug are increased significantly when it is taken with food.[8]
Distribution
Doxepin is widely distributed throughout the body and is approximately 80% plasma protein-bound, specifically to albumin and α1-acid glycoprotein.[8][69]
Metabolism
Doxepin is extensively metabolized by the liver via oxidation and N-demethylation.[8] Its metabolism is highly stereoselective.[70] Based on in vitro research, the major enzymes involved in the metabolism of doxepin are the cytochrome P450 enzymes CYP2D6 and CYP2C19, with CYP1A2, CYP2C9, and CYP3A4 also involved to a lesser extent.[8][70] The major active metabolite of doxepin, nordoxepin, is formed mainly by CYP2C19 (>50% contribution), while CYP1A2 and CYP2C9 are involved to a lesser extent, and CYP2D6 and CYP3A4 are not involved.[71] Both doxepin and nordoxepin are hydroxylated mainly by CYP2D6,[72] and both doxepin and nordoxepin are also transformed into glucuronide conjugates.[48][8] The elimination half-life of doxepin is about 15–18 hours, whereas that of nordoxepin is around 28–31 hours.[8][9] Up to 10% of Caucasian individuals show substantially reduced metabolism of doxepin that can result in up to 8-fold elevated plasma concentrations of the drug compared to normal.[49][48]
Nordoxepin is a mixture of (E) and (Z) stereoisomers similarly to doxepin.[5] Whereas pharmaceutical doxepin is supplied in an approximate 85:15 ratio mixture of (E)- and (Z)-stereoisomers and plasma concentrations of doxepin remain roughly the same as this ratio with treatment, plasma levels of the (E)- and (Z)-stereoisomers of nordoxepin, due to stereoselective metabolism of doxepin by cytochrome P450 enzymes, are approximately 1:1.[5]
Elimination
Doxepin is excreted primarily in the urine and predominantly in the form of glucuronide conjugates, with less than 3% of a dose excreted unchanged as doxepin or nordoxepin.[8]
Pharmacogenetics
Since doxepin is mainly metabolized by CYP2D6, CYP2C9, and CYP2C19, genetic variations within the genes coding for these enzymes can affect its metabolism, leading to changes in the concentrations of the drug in the body. Increased concentrations of doxepin may increase the risk for side effects, including anticholinergic and nervous system adverse effects, while decreased concentrations may reduce the drug's efficacy.
Individuals can be categorized into different types of cytochrome P450 metabolizers depending on which genetic variations they carry. These metabolizer types include poor, intermediate, extensive, and ultrarapid metabolizers. Most people are extensive metabolizers, and have "normal" metabolism of doxepin. Poor and intermediate metabolizers have reduced metabolism of the drug as compared to extensive metabolizers; patients with these metabolizer types may have an increased probability of experiencing side effects. Ultrarapid metabolizers break down doxepin much faster than extensive metabolizers; patients with this metabolizer type may have a greater chance of experiencing pharmacological failure.
A study assessed the metabolism of a single 75 mg oral dose of doxepin in healthy volunteers with genetic polymorphisms in CYP2D6, CYP2C9, and CYP2C19 enzymes.[70] In CYP2D6 extensive, intermediate, and poor metabolizers, the mean clearance rates of (E)-doxepin were 406, 247, and 127 L/hour, respectively (~3-fold difference between extensive and poor).[70] In addition, the bioavailability of (E)-doxepin was about 2-fold lower in extensive relative to poor CYP2D6 metabolizers, indicating a significant role of CYP2D6 in the first-pass metabolism of (E)-doxepin.[70] The clearance of (E)-doxepin in CYP2C9 slow metabolizers was also significantly reduced at 238 L/hour.[70] CYP2C19 was involved in the metabolism of (Z)-doxepin, with clearance rates of 191 L/hour in CYP2C19 extensive metabolizers and 73 L/hour in poor metabolizers (~2.5-fold difference).[70] Area-under-the-curve (0–48 hour) levels of nordoxepin were dependent on the genotype of CYP2D6 with median values of 1.28, 1.35, and 5.28 nM•L/hour in CYP2D6 extensive, intermediate, and poor metabolizers, respectively (~4-fold difference between extensive and poor).[70] Taken together, doxepin metabolism appears to be highly stereoselective, and CYP2D6 genotype has a major influence on the pharmacokinetics of (E)-doxepin.[70] Moreover, CYP2D6 poor metabolizers, as well as patients taking potent CYP2D6 inhibitors (which can potentially convert a CYP2D6 extensive metabolizer into a poor metabolizer), may be at an increased risk for adverse effects of doxepin due to their slower clearance of the drug.[70]
Another study assessed doxepin and nordoxepin metabolism in CYP2D6 ultra-rapid, extensive, and poor metabolizers following a single 75 mg oral dose.[72] They found up to more than 10-fold variation in total exposure to doxepin and nordoxepin between the different groups.[72] The researchers suggested that in order to achieve equivalent exposure, based on an average dose of 100%, the dosage of doxepin might be adjusted to 250% in ultra-rapid metabolizers, 150% in extensive metabolizers, 50% in intermediate metabolizers, and 30% in poor metabolizers.[72]
Chemistry
Doxepin is a tricyclic compound, specifically a dibenzoxepin, and possesses three rings fused together with a side chain attached in its chemical structure.[48] It is the only TCA with a dibenzoxepin ring system to have been marketed.[73] Doxepin is a tertiary amine TCA, with its side chain-demethylated metabolite nordoxepin being a secondary amine.[50][51] Other tertiary amine TCAs include amitriptyline, imipramine, clomipramine, dosulepin (dothiepin), and trimipramine.[74][75] Doxepin is a mixture of (E) and (Z) stereoisomers (the latter being known as cidoxepin or cis-doxepin) and is used commercially in a ratio of approximately 85:15.[2][76] The chemical name of doxepin is (E/Z)-3-(dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine[48][77] and its free base form has a chemical formula of C19H21NO with a molecular weight of 279.376 g/mol.[77] The drug is used commercially almost exclusively as the hydrochloride salt; the free base has been used rarely.[2][78] The CAS Registry Number of the free base is 1668-19-5 and of the hydrochloride is 1229-29-4.[2][78]
History
Doxepin was discovered in Germany in 1963 and was introduced in the United States as an antidepressant in 1969.[48] It was subsequently approved at very low doses in the United States for the treatment of insomnia in 2010.[54][78]
Society and culture
Generic names
Doxepin is the generic name of the drug in English and German and its INNTooltip International Nonproprietary Name and BANTooltip British Approved Name, while doxepin hydrochloride is its USANTooltip United States Adopted Name, USPTooltip United States Pharmacopeia, BANMTooltip British Approved Name, and JANTooltip Japanese Accepted Name.[2][78][79][1] Its generic name in Spanish and Italian and its DCITTooltip Denominazione Comune Italiana are doxepina, in French and its DCFTooltip Dénomination Commune Française are doxépine, and in Latin is doxepinum.[1]
The cis or (Z) stereoisomer of doxepin is known as cidoxepin, and this is its INNTooltip International Nonproprietary Name while cidoxepin hydrochloride is its USANTooltip United States Adopted Name.[2]
Brand names
It was introduced under the brand names Quitaxon and Aponal by Boehringer and as Sinequan by Pfizer.[80]
Doxepin is marketed under many brand names worldwide, including: Adnor, Anten, Antidoxe, Colian, Deptran, Dofu, Doneurin, Dospin, Doxal, Doxepini, Doxesom, Doxiderm, Flake, Gilex, Ichderm, Li Ke Ning, Mareen, Noctaderm, Oxpin, Patoderm, Prudoxin, Qualiquan, Quitaxon, Sagalon, Silenor, Sinepin, Sinequan, Sinquan, and Zonalon.[1] It is also marketed as a combination drug with levomenthol under the brand name Doxure.[1]
Approvals
The oral formulations of doxepin are FDATooltip Food and Drug Administration-approved for the treatment of depression and sleep-maintenance insomnia, and its topical formulations are FDA-approved the short-term management for some itchy skin conditions.[81] In Australia and the United Kingdom, the only licensed indications are in the treatment of major depression and pruritus in eczema.[30][82]
Research
Antihistamine
Cidoxepin is under development by Elorac, Inc. for the treatment of chronic urticaria (hives).[83] As of 2017, it is in phase II clinical trials for this indication.[83] The drug was also under investigation for the treatment of allergic rhinitis, atopic dermatitis, and contact dermatitis, but development for these indications was discontinued.[83]
Headache
Doxepin was under development by Winston Pharmaceuticals in an intranasal formulation for the treatment of headache.[84] As of August 2015, it was in phase II clinical trials for this indication.[84]
Neuropathic pain
As of 2017, there was no good evidence that topical doxepin was useful to treat localized neuropathic pain.[85]
References
- "International brands of doxepin". Drugs.com. Retrieved 25 October 2017.
- Elks J (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 469–. ISBN 978-1-4757-2085-3.
- "Doxepin Hydrochloride". Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. 30 January 2013. Retrieved 3 December 2013.
- Anvisa (2023-03-31). "RDC Nº 784 - Listas de Substâncias Entorpecentes, Psicotrópicas, Precursoras e Outras sob Controle Especial" [Collegiate Board Resolution No. 784 - Lists of Narcotic, Psychotropic, Precursor, and Other Substances under Special Control] (in Brazilian Portuguese). Diário Oficial da União (published 2023-04-04). Archived from the original on 2023-08-03. Retrieved 2023-08-16.
- Thomas L. Lemke; David A. Williams (24 January 2012). Foye's Principles of Medicinal Chemistry. Lippincott Williams & Wilkins. pp. 604–. ISBN 978-1-60913-345-0.
- Yan JH, Hubbard JW, McKay G, Korchinski ED, Midha KK (July 2002). "Absolute bioavailability and stereoselective pharmacokinetics of doxepin". Xenobiotica; the Fate of Foreign Compounds in Biological Systems. 32 (7): 615–23. doi:10.1080/00498250210131879. PMID 12162857. S2CID 7400543.
- "Sinepin Capsules 25mg - Summary of Product Characteristics (SPC)". UK Electronic Medicines Compendium. 22 January 2014. Retrieved 24 October 2017.
- Weber J, Siddiqui MA, Wagstaff AJ, McCormack PL (August 2010). "Low-dose doxepin: in the treatment of insomnia". CNS Drugs. 24 (8): 713–20. doi:10.2165/11200810-000000000-00000. PMID 20658801. S2CID 26739281.
- Virtanen R, Scheinin M, Iisalo E (November 1980). "Single dose pharmacokinetics of doxepin in healthy volunteers". Acta Pharmacologica et Toxicologica. 47 (5): 371–6. doi:10.1111/j.1600-0773.1980.tb01575.x. PMID 7293791.
- "Doxepin Hydrochloride". Drugs.com. American Society of Health-System Pharmacists. Retrieved 20 March 2019.
- "Silenor (doxepin) label" (PDF). FDA. 17 March 2010. Retrieved 25 October 2017. For label updates see FDA index page for NDA 022036
- Yeung WF, Chung KF, Yung KP, Ng TH (February 2015). "Doxepin for insomnia: a systematic review of randomized placebo-controlled trials". Sleep Medicine Reviews. 19: 75–83. doi:10.1016/j.smrv.2014.06.001. PMID 25047681.
- "Doxepin hydrochloride cream" (PDF). FDA. 20 December 2002. Retrieved 25 October 2017. For label updates see FDA index page for NDA 020126
- British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. p. 372. ISBN 9780857113382.
- "Doxepin Use During Pregnancy". Drugs.com. Retrieved 21 March 2019.
- "Competitive Generic Therapy Approvals". U.S. Food and Drug Administration (FDA). 29 June 2023. Archived from the original on 29 June 2023. Retrieved 29 June 2023.
- "First Generic Drug Approvals 2023". U.S. Food and Drug Administration (FDA). 30 May 2023. Archived from the original on 30 June 2023. Retrieved 30 June 2023.
- "The Top 300 of 2020". ClinCalc. Retrieved 7 October 2022.
- "Doxepin - Drug Usage Statistics". ClinCalc. Retrieved 7 October 2022.
- Sateia MJ, Buysse DJ, Krystal AD, Neubauer DN, Heald JL (February 2017). "Clinical Practice Guideline for the Pharmacologic Treatment of Chronic Insomnia in Adults: An American Academy of Sleep Medicine Clinical Practice Guideline". Journal of Clinical Sleep Medicine. 13 (2): 307–349. doi:10.5664/jcsm.6470. PMC 5263087. PMID 27998379.
- Qaseem A, Kansagara D, Forciea MA, Cooke M, Denberg TD (July 2016). "Management of Chronic Insomnia Disorder in Adults: A Clinical Practice Guideline From the American College of Physicians". Annals of Internal Medicine. 165 (2): 125–33. doi:10.7326/M15-2175. PMID 27136449.
- Brasure M, MacDonald R, Fuchs E, Olson CM, Carlyle M, Diem S, et al. (2015). "Executive Summary". Management of Insomnia Disorder: Executive Summary. PMID 26844312.
{{cite book}}:|journal=ignored (help) - De Crescenzo F, D'Alò GL, Ostinelli EG, Ciabattini M, Di Franco V, Watanabe N, Kurtulmus A, Tomlinson A, Mitrova Z, Foti F, Del Giovane C, Quested DJ, Cowen PJ, Barbui C, Amato L, Efthimiou O, Cipriani A (July 2022). "Comparative effects of pharmacological interventions for the acute and long-term management of insomnia disorder in adults: a systematic review and network meta-analysis". Lancet. 400 (10347): 170–184. doi:10.1016/S0140-6736(22)00878-9. PMID 35843245. S2CID 250536370.
- Dupuis G, Vaugeois JM (February 2020). "Les effets anti-H1 intéressants dans les insomnies de maintien : réflexion sur les intérêts comparés de la doxylamine et de la doxépine" [The interesting anti-H1 effects in maintenance insomnia: A reflection on the comparative advantages of doxylamine and doxepin]. Encephale (in French). 46 (1): 80–82. doi:10.1016/j.encep.2019.01.006. PMID 30879783. S2CID 151085176.
- Everitt H, Baldwin DS, Stuart B, Lipinska G, Mayers A, Malizia AL, Manson CC, Wilson S (May 2018). "Antidepressants for insomnia in adults". Cochrane Database Syst Rev. 2018 (5): CD010753. doi:10.1002/14651858.CD010753.pub2. PMC 6494576. PMID 29761479.
- Eschler DC, Klein PA (August 2010). "An evidence-based review of the efficacy of topical antihistamines in the relief of pruritus". Journal of Drugs in Dermatology. 9 (8): 992–7. PMID 20684150.
- Fernando S, Broadfoot A (March 2010). "Chronic urticaria--assessment and treatment" (PDF). Australian Family Physician. 39 (3): 135–8. PMID 20369115.
- "Deptran Doxepin (as hydrochloride)" (PDF). TGA eBusiness Services. Alphapharm Pty Ltd. 6 May 2013. Retrieved 3 December 2013.
- "Silenor (doxepin) dosing, indications, interactions, adverse effects, and more". Medscape Reference. WebMD. Retrieved 3 December 2013.
- Rossi, S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- Lippincott "nursing 2007 drug handbook" LWW press. 2007
- White N, Litovitz T, Clancy C (December 2008). "Suicidal antidepressant overdoses: a comparative analysis by antidepressant type". Journal of Medical Toxicology. 4 (4): 238–50. doi:10.1007/BF03161207. PMC 3550116. PMID 19031375.
- Myers B, Reddy V, Chan S, Thibodeaux Q, Brownstone N, Koo J (February 2022). "Optimizing doxepin therapy in dermatology: introducing blood level monitoring and genotype testing". The Journal of Dermatological Treatment. 33 (1): 87–93. doi:10.1080/09546634.2020.1762841. PMID 32347140. S2CID 216647836.
- Krystal AD, Richelson E, Roth T (August 2013). "Review of the histamine system and the clinical effects of H1 antagonists: basis for a new model for understanding the effects of insomnia medications". Sleep Medicine Reviews. 17 (4): 263–72. doi:10.1016/j.smrv.2012.08.001. PMID 23357028.
- Roth, BL; Driscol, J. "PDSP Ki Database". Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
- Tatsumi M, Groshan K, Blakely RD, Richelson E (December 1997). "Pharmacological profile of antidepressants and related compounds at human monoamine transporters". European Journal of Pharmacology. 340 (2–3): 249–58. doi:10.1016/s0014-2999(97)01393-9. PMID 9537821.
- Cusack B, Nelson A, Richelson E (May 1994). "Binding of antidepressants to human brain receptors: focus on newer generation compounds". Psychopharmacology. 114 (4): 559–65. doi:10.1007/bf02244985. PMID 7855217. S2CID 21236268.
- Pälvimäki EP, Roth BL, Majasuo H, Laakso A, Kuoppamäki M, Syvälahti E, Hietala J (August 1996). "Interactions of selective serotonin reuptake inhibitors with the serotonin 5-HT2c receptor". Psychopharmacology. 126 (3): 234–40. doi:10.1007/bf02246453. PMID 8876023. S2CID 24889381.
- Gumilar F, Bouzat C (April 2008). "Tricyclic antidepressants inhibit homomeric Cys-loop receptors by acting at different conformational states". European Journal of Pharmacology. 584 (1): 30–9. doi:10.1016/j.ejphar.2008.01.023. hdl:11336/44466. PMID 18314100.
- Monsma FJ, Shen Y, Ward RP, Hamblin MW, Sibley DR (March 1993). "Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs". Molecular Pharmacology. 43 (3): 320–7. PMID 7680751.
- Appl H, Holzammer T, Dove S, Haen E, Strasser A, Seifert R (February 2012). "Interactions of recombinant human histamine H1R, H2R, H3R, and H4R receptors with 34 antidepressants and antipsychotics". Naunyn-Schmiedeberg's Archives of Pharmacology. 385 (2): 145–70. doi:10.1007/s00210-011-0704-0. PMID 22033803. S2CID 14274150.
- Nguyen T, Shapiro DA, George SR, Setola V, Lee DK, Cheng R, et al. (March 2001). "Discovery of a novel member of the histamine receptor family". Molecular Pharmacology. 59 (3): 427–33. doi:10.1124/mol.59.3.427. PMID 11179435.
- Richelson E, Nelson A (July 1984). "Antagonism by antidepressants of neurotransmitter receptors of normal human brain in vitro". The Journal of Pharmacology and Experimental Therapeutics. 230 (1): 94–102. PMID 6086881.
- Stanton T, Bolden-Watson C, Cusack B, Richelson E (June 1993). "Antagonism of the five cloned human muscarinic cholinergic receptors expressed in CHO-K1 cells by antidepressants and antihistaminics". Biochemical Pharmacology. 45 (11): 2352–4. doi:10.1016/0006-2952(93)90211-e. PMID 8100134.
- Duncan RS, McPate MJ, Ridley JM, Gao Z, James AF, Leishman DJ, et al. (August 2007). "Inhibition of the HERG potassium channel by the tricyclic antidepressant doxepin". Biochemical Pharmacology. 74 (3): 425–37. doi:10.1016/j.bcp.2007.04.024. PMC 1920586. PMID 17560554.
- Thanacoody HK, Thomas SH (2005). "Tricyclic antidepressant poisoning : cardiovascular toxicity". Toxicological Reviews. 24 (3): 205–14. doi:10.2165/00139709-200524030-00013. PMID 16390222. S2CID 44532041.
- Bertelsen AK, Backonja MM (2007). "Drugs Targeting Voltage-Gated Sodium and Calcium Channels". Encyclopedia of Pain. p. 651. doi:10.1007/978-3-540-29805-2_1205. ISBN 978-3-540-43957-8.
- Singh H, Becker PM (August 2007). "Novel therapeutic usage of low-dose doxepin hydrochloride". Expert Opinion on Investigational Drugs. 16 (8): 1295–305. doi:10.1517/13543784.16.8.1295. PMID 17685877. S2CID 32810608.
- Lankford A (2011). "Low-dose doxepin (3 and 6 mg) for the treatment of insomnia". Future Neurology. 6 (2): 143–154. doi:10.2217/fnl.10.83. ISSN 1479-6708.
- Neal R. Cutler; John J. Sramek; Prem K. Narang (20 September 1994). Pharmacodynamics and Drug Development: Perspectives in Clinical Pharmacology. John Wiley & Sons. pp. 160–. ISBN 978-0-471-95052-3.
- Pavel Anzenbacher; Ulrich M. Zanger (23 February 2012). Metabolism of Drugs and Other Xenobiotics. John Wiley & Sons. pp. 302–. ISBN 978-3-527-64632-6.
- Alan F. Schatzberg; Charles B. Nemeroff (2009). The American Psychiatric Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 264–. ISBN 978-1-58562-309-9.
- Rojas-Fernandez CH, Chen Y (September 2014). "Use of ultra-low-dose (≤6 mg) doxepin for treatment of insomnia in older people". Canadian Pharmacists Journal. 147 (5): 281–9. doi:10.1177/1715163514543856. PMC 4213269. PMID 25364337.
- Yeung WF, Chung KF, Yung KP, Ng TH (February 2015). "Doxepin for insomnia: a systematic review of randomized placebo-controlled trials". Sleep Medicine Reviews. 19: 75–83. doi:10.1016/j.smrv.2014.06.001. PMID 25047681.
- Gillman PK (July 2007). "Tricyclic antidepressant pharmacology and therapeutic drug interactions updated". British Journal of Pharmacology. 151 (6): 737–48. doi:10.1038/sj.bjp.0707253. PMC 2014120. PMID 17471183.
- Tariq SH, Pulisetty S (February 2008). "Pharmacotherapy for insomnia". Clinics in Geriatric Medicine. 24 (1): 93–105, vii. doi:10.1016/j.cger.2007.08.009. PMID 18035234.
- Laurence Brunton; Bruce A. Chabner; Bjorn Knollman (14 January 2011). Goodman and Gilman's The Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional. p. 410. ISBN 978-0-07-176939-6.
- Stahl SM (December 2008). "Selective histamine H1 antagonism: novel hypnotic and pharmacologic actions challenge classical notions of antihistamines". CNS Spectrums. 13 (12): 1027–38. doi:10.1017/s1092852900017089. PMID 19179941. S2CID 6849261.
- Richelson E (October 1979). "Tricyclic antidepressants and histamine H1 receptors". Mayo Clinic Proceedings. 54 (10): 669–74. PMID 39202.
- Alan F. Schatzberg; Charles B. Nemeroff (10 May 2017). The American Psychiatric Association Publishing Textbook of Psychopharmacology. American Psychiatric Pub. pp. 322–. ISBN 978-1-61537-122-8.
- Stahl SM (February 2009). "Multifunctional drugs: a novel concept for psychopharmacology". CNS Spectrums. 14 (2): 71–3. doi:10.1017/s1092852900000213. PMID 19238121. S2CID 2587534.
- Vande Griend JP, Anderson SL (2012). "Histamine-1 receptor antagonism for treatment of insomnia". Journal of the American Pharmacists Association. 52 (6): e210-9. doi:10.1331/JAPhA.2012.12051. PMID 23229983.
- "Sleep Disorder (Sedative-Hypnotic) Drug Information - U.S. FDA". Food and Drug Administration. 13 June 2017. Retrieved 9 August 2017.
- Mets MA, van Deventer KR, Olivier B, Verster JC (2013). "Critical appraisal of ramelteon in the treatment of insomnia". Nature and Science of Sleep. 2 (4): 257–66. doi:10.1016/S2222-1808(13)60080-8. PMC 4027305.
In general, sedating properties of anti-depressant agents are related to antagonism of serotonin 5HT2, histamines, and α-1 adrenergic receptors[14]–[16].
- Landolt HP, Wehrle R (May 2009). "Antagonism of serotonergic 5-HT2A/2C receptors: mutual improvement of sleep, cognition and mood?". The European Journal of Neuroscience. 29 (9): 1795–809. doi:10.1111/j.1460-9568.2009.06718.x. PMID 19473234. S2CID 17097545.
- Broese M, Riemann D, Hein L, Nissen C (September 2012). "α-Adrenergic receptor function, arousal and sleep: mechanisms and therapeutic implications". Pharmacopsychiatry. 45 (6): 209–16. doi:10.1055/s-0031-1299728. PMID 22290201. S2CID 28763568.
- Szewczuk-Bogusławska M, Kiejna A, Beszłej JA, Orzechowska-Juzwenko K, Milejski P (2004). "Doxepin inhibits CYP2D6 activity in vivo". Polish Journal of Pharmacology. 56 (4): 491–4. PMID 15520506.
- Leucht S, Steimer W, Kreuz S, Abraham D, Orsulak PJ, Kissling W (August 2001). "Doxepin plasma concentrations: is there really a therapeutic range?". Journal of Clinical Psychopharmacology. 21 (4): 432–9. doi:10.1097/00004714-200108000-00011. PMID 11476128. S2CID 32147467.
- Virtanen R, Iisalo E, Irjala K (August 1982). "Protein binding of doxepin and desmethyldoxepin". Acta Pharmacologica et Toxicologica. 51 (2): 159–64. doi:10.1111/j.1600-0773.1982.tb01008.x. PMID 7113722.
- Kirchheiner J, Meineke I, Müller G, Roots I, Brockmöller J (October 2002). "Contributions of CYP2D6, CYP2C9 and CYP2C19 to the biotransformation of E- and Z-doxepin in healthy volunteers". Pharmacogenetics. 12 (7): 571–80. doi:10.1097/00008571-200210000-00010. PMID 12360109.
- Härtter S, Tybring G, Friedberg T, Weigmann H, Hiemke C (July 2002). "The N-demethylation of the doxepin isomers is mainly catalyzed by the polymorphic CYP2C19". Pharmaceutical Research. 19 (7): 1034–7. doi:10.1023/a:1016478708902. PMID 12180536. S2CID 8089917.
- Kirchheiner J, Henckel HB, Franke L, Meineke I, Tzvetkov M, Uebelhack R, et al. (August 2005). "Impact of the CYP2D6 ultra-rapid metabolizer genotype on doxepin pharmacokinetics and serotonin in platelets". Pharmacogenetics and Genomics. 15 (8): 579–87. doi:10.1097/01.fpc.0000167331.30905.9e. PMID 16007002. S2CID 41765748.
- Manuchair Ebadi (31 October 2007). Desk Reference of Clinical Pharmacology, Second Edition. CRC Press. pp. 329–. ISBN 978-1-4200-4744-8.
- Patricia K. Anthony (2002). Pharmacology Secrets. Elsevier Health Sciences. pp. 39–. ISBN 1-56053-470-2.
- Philip Cowen; Paul Harrison; Tom Burns (9 August 2012). Shorter Oxford Textbook of Psychiatry. OUP Oxford. pp. 532–. ISBN 978-0-19-162675-3.
- Shufeng Zhou (6 April 2016). Cytochrome P450 2D6: Structure, Function, Regulation and Polymorphism. CRC Press. pp. 142–. ISBN 978-1-4665-9788-4.
- Chambers M. "Doxepin [INN:BAN] - Similar structures search, synonyms, formulas, resource links, and other chemical information". ChemIDplus. U.S. National Library of Medicine. Retrieved 16 March 2019.
- Index Nominum 2000: International Drug Directory. Taylor & Francis. 2000. pp. 370–. ISBN 978-3-88763-075-1.
- I.K. Morton; Judith M. Hall (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 106–. ISBN 978-94-011-4439-1.
- Adis Editorial (1971). "Doxepin". Drugs. 1 (3): 194–227. doi:10.2165/00003495-197101030-00002. S2CID 46963857.
- "PRUDOXIN (doxepin hydrochloride) cream". DailyMed. August 2010. Retrieved 3 December 2013.
- Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- "Cidoxepin". Adisinsight.springer.com. Retrieved 16 March 2019.
- "Doxepin intranasal - Winston Pharmaceuticals". Adisinsight.springer.com. Retrieved 16 March 2019.
- Casale R, Symeonidou Z, Bartolo M (March 2017). "Topical Treatments for Localized Neuropathic Pain". Current Pain and Headache Reports. 21 (3): 15. doi:10.1007/s11916-017-0615-y. PMC 5340828. PMID 28271334.
External links
![]() Media related to Doxepin at Wikimedia Commons
Media related to Doxepin at Wikimedia Commons
- "Doxepin". Drug Information Portal. U.S. National Library of Medicine.
- "Doxepin hydrochloride". Drug Information Portal. U.S. National Library of Medicine.