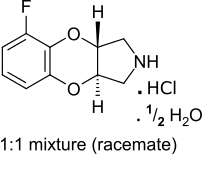
Fluparoxan
Fluparoxan (developmental code name GR50360A) is a potent α2-adrenergic receptor antagonist (pKB = 7.9) with excellent selectivity for this receptor over the α1-adrenergic receptor (2,630-fold), and is the only well-studied α2-adrenergic receptor antagonist in its structural family which does not antagonize any variant of the imidazoline receptor.[1][2] It was shown to possess central α2-adrenoceptor antagonist activity after oral doses in man and was patented as an antidepressant by Glaxo in the early 1980s, but its development was discontinued when the compound failed to show a clear clinical advantage over existing therapies.[1]
 | |
| Clinical data | |
|---|---|
| Other names | (+/-)-(trans)-5-fluoro-2,3,3a,9a-tetrahydro-1H-[1,4]benzodioxino[2,3-c]pyrrole hydrochloride hemihydrate |
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 85% oral from tablet formulation |
| Metabolism | Greater than 90% excreted as (sulphamic acid and carbamoyI glucuronide conjugates) |
| Elimination half-life | 6 hours |
| Excretion | Renal |
| Identifiers | |
IUPAC name
| |
| CAS Number |
|
| UNII |
|
| CompTox Dashboard (EPA) |
|
| Chemical and physical data | |
| Formula | C10H12ClFNO2.5 |
| Molar mass | 240.66 g·mol−1 |
| 3D model (JSmol) |
|
SMILES
| |
Pharmacology
Pharmacodynamics
Fluparoxan is a very selective α2-adrenergic blocker,[3] that readily crosses the blood–brain barrier. Blockade of α2-adrenoreceptors, particularly presynaptic autoreceptors in noradrenergic neurons by fluparoxan, produces an increase in the synaptic concentrations of noradrenaline,[4] by blocking the autoinhibitory feedback mechanism. This release of noradrenaline has a potential value in the treatment of disorders which are associated with a deficiency of noradrenaline at postsynaptic adrenoreceptors, such as depression,[5] the early features of Alzheimer's disease and schizophrenia and other neurodevelopmental disorders associated with cognitive impairment.[6][7] Fluparoxan also exhibits no anticholinergic, antidopaminergic, α1-adrenergic, β-adrenergic, muscarinic, or 5-HT1 receptor-blocking effects.[3]
Fluparoxan showed α2-adrenoceptors antagonist activity in vivo in several animal species.[3] In the conscious mouse, fluparoxan was effective by the oral route in preventing clonidine-induced hypothermia and antinociception. While in the rat the marked impairment of rotarod performance were prevented dose-dependently by fluparoxan. Fluparoxan, orally prevented agonist UK-14304 induced sedation and bradycardia in a dose-related fashion in the dog. Fluparoxan has been shown to possess central α2-adrenoceptor antagonist activity after both single and repeated oral doses in man, significantly attenuating all responses to the agonist clonidine (growth hormone secretion, bradycardia, hypotension, xerostomia) apart from the measures of sedation.[8]
Fluparoxan has shown positive effects in the treatment of cognitive dysfunction in schizophrenia patients when orally dosed with fluparoxan,[1] and in the treatment of central neurodegenerative disorders in models of Alzheimer's disease where it prevented age-related decline in spatial working memory in transgenic mice, although it had no effect in other memory tasks such as object recognition or the Morris water maze and occurred in the absence of obvious concomitant change in pathology such as β-amyloid plaque load and astrocytosis.[9]
The individual chiral isomers 7 (+) 3aR,9aR and 8 (-) 3aS,9aS of fluparoxan both had comparable levels of α2-adrenoceptors antagonist potency and α2/α1 selectivity to racemic fluparoxan 6 (±) rel 3aR,9aR in vitro and a similar potency in vivo in reversing the hypothermia induced in mice by clonidine.[3]
Enantiomer_and_(-)Enantiomer.svg.png.webp)
| Compound as HCl | α2-Adrenoceptors Vas deferens UK14304 | α1-Adrenoceptors Anococcygeus muscle Phenylephrine | Hypothermia Clonidine Mice po | Optical Rotation | Melting Point |
|---|---|---|---|---|---|
| Measurement | pKB | pKB | ED50mg/kg | [α]20-23D° | mp°C |
| 6 (+/-) rel 3aR,9aR | 7.86 | 5.00 | 0.66 | 0 | 245 |
| 7 (+) 3aR,9aR | 7.88 | 4.47 | 0.73 | +159.6 | 235-236 |
| 8 (-) 3aS,9aS | 7.68 | 4.95 | 0.46 | -159.4 | 238-240 |
Pharmacokinetics
Fluparoxan has rat and human protein binding of 81-92% and 95% respectively. Fluparoxan shows high in vitro permeability in MCDK (Papp nm/s =2500) and Caco-2 (Papp nm/s =2000) cells which correlates well with the known high oral intestinal absorption (100%) in humans.[10] Fluparoxan is well absorbed following oral dosing in all animals. Clearance was largely metabolic, with both oral and intravenous doses being excreted mainly via the urine (> 90%, of administered dose), chiefly as phase II metabolites (sulphamic acid and carbamoyI glucuronide conjugates).[11] Fluparoxan has high oral bioavailability (100%) and a long duration of action (2hr) in the rat,[1] which accounts for its similar potency seen by both the oral and intravenous routes of administration in this species.[3] The excellent pharmacokinetics exhibited by fluparoxan in animals also translated into man [1] where it has a superior bioavailability (97%) and longer duration of action (6-7hrs).
Metabolism
Fluparoxan was not metabolized by human cytochrome P450 enzymes CYP1A and CYP2A, and was found not to be a mutagen in cultured human peripheral lymphocytes and did not cause gene mutation when administered to Chinese hamster fibroblasts in culture.[11] No mutagenic potential was identified in microbiological mutagenicity tests including a fluctuation test with S9 activation. No hydroxylated metabolites were identified after incubation with rat liver microsomes (S9) or in rat urine following oral dosing.[1] The compound was well tolerated on repeat oral administration to rat (≤ 200 mg /kg/day) and dog (≤ 80 mg/kg/day) for 12 months.
Chemistry
Racemic (±) fluparoxan 6 is a white crystalline powder as the hydrochloride hemihydrate m.p. 245 °C.[1] It is a mixture of two enantiomers and is moderately lipophilic (log P = 1.2) with good solubility 80 mg/mL in water at 25 °C. The compound is very stable in the solid state and its bioavailability from tablet formulation is 85% and absorption is rapid.[1]
Synthesis
Racemic (±) fluparoxan 6 was first synthesised by a convergent route,[1][2] which involved converting the bis-benzyl ether of cis-butene-l,4-diol 1 into its epoxide 2 followed by acid catalyzed ring opening to a racemic-diol which was then converted into the racemic trans bis-tosylate 3. Coupling 3-fluorocatechol 4 with the racemic trans bis-tosylate 3 in the presence base gave on deprotection the benzodioxan diol 5 which was converted to racemic 6 via coupling its bis-mesylate to benzylamine followed by deprotection.[1][2]

The individual chiral isomers (+) 7 and (-) 8 of fluparoxan were originally prepared from the (+) and (-) isomers of diethyl tartrate via the corresponding chiral trans bis-tosylates in a convergent route similar to that shown in the synthesis of racemic fluparoxan.[1]
History
Although fluparoxan has been shown to possess central α2-adrenoceptor antagonist activity after both single and repeated oral doses in man, clinical evaluation in depression and its development for the treatment of male sexual dysfunction was discontinued in the early 1990s when the compound failed to show a clear clinical advantage over existing therapies. The same is true for other α2-adrenoceptor antagonists in depression and it is now generally agreed that the original hypothesis that blocking α2-presynaptic adrenoceptors to increase brain levels of noradrenalin is insufficient as a neurobiological basis for depressive disorders, with the true picture likely to be much more complex and heterogeneous, involving both monoaminergic and nonmonoaminergic players.[12] In contrast however, recent interest in fluparoxan has increased with its positive effects in treating cognitive dysfunction in central neurodegenerative diseases.[1]
See also
- Efaroxan
- Idazoxan
References
- Borthwick AD (2017). "Fluparoxan: A Comprehensive Review of its Discovery, Adrenergic and CNS activity and treatment of Cognitive Dysfunction in central Neurodegenerative diseases". Mini Reviews in Medicinal Chemistry. 17 (7): 572–582. doi:10.2174/1389557516666160321115041. PMID 26996616.
- Kitchin J, Borthwick AD, Brodie AC, Cherry PC, Crame AJ, Pipe AJ, Procopiou PA, Seaman MA, Turnbull JP (December 1995). "Synthesis of Benzodioxinopyrroles as selective α 2-adrenoceptor antagonists". Bioorganic & Medicinal Chemistry. 3 (12): 1595–1603. doi:10.1016/0968-0896(95)00143-3. PMID 8770384.
- Halliday CA, Jones BJ, Skingle M, Walsh DM, Wise H, Tyers MB (April 1991). "The pharmacology of fluparoxan: a selective α2‐adrenoceptor antagonist". British Journal of Pharmacology. 102 (4): 887–895. doi:10.1111/j.1476-5381.1991.tb12272.x. PMC 1917968. PMID 1677298.
- Fernández-Pastor B, Meana JJ (May 2002). "In vivo tonic modulation of the noradrenaline release in the rat cortex by locus coeruleus somatodendritic α2-adrenoceptors". European Journal of Pharmacology. 442 (3): 225–229. doi:10.1016/S0014-2999(02)01543-1. PMID 12065075.
- Cottingham C, Wang Q (November 2012). "α2 Adrenergic receptor dysregulation in depressive disorders: implications for the neurobiology of depression and antidepressant therapy". Neuroscience & Biobehavioral Reviews. 36 (10): 2214–2225. doi:10.1016/j.neubiorev.2012.07.011. PMC 3508310. PMID 22910678.
- Marien MR, Colpaert FC, Rosenquist AC (April 2004). "Noradrenergic mechanisms in neurodegenerative diseases: a theory". Brain Research Reviews. 45 (1): 38–78. doi:10.1016/j.brainresrev.2004.02.002. PMID 15063099. S2CID 20549357.
- Gibbs AA, Naudts KH, Azevedo RT, David AS (April 2010). "Deletion variant of α2b-adrenergic receptor gene moderates the effect of COMT val 158 met polymorphism on episodic memory performance". European Neuropsychopharmacology. 20 (4): 272–275. doi:10.1016/j.euroneuro.2009.12.007. PMID 20110158. S2CID 32471274.
- Johnson MA, Blackwell CP, Smith J (May 1995). "Antagonism of the effects of clonidine by the alpha 2‐adrenoceptor antagonist, fluparoxan". British Journal of Clinical Pharmacology. 39 (5): 477–483. doi:10.1111/j.1365-2125.1995.tb04483.x. PMC 1365053. PMID 7669482.
- Scullion GA, Kendall DA, Marsden CA, Sunter D, Pardon MC (March 2011). "Chronic treatment with the α 2-adrenoceptor antagonist fluparoxan prevents age-related deficits in spatial working memory in APP× PS1 transgenic mice without altering β-amyloid plaque load or astrocytosis". Neuropharmacology. 60 (2): 223–234. doi:10.1016/j.neuropharm.2010.09.002. PMID 20850464. S2CID 32320290.
- Gristwood WE (December 1990). "Determination of fluparoxan (GR50360) in plasma by gas chromatography". Journal of Chromatography B. 527 (2): 436–440. doi:10.1016/S0378-4347(00)82128-3. PMID 2387889.
- Beresford AP, Ellis WJ, Ayrton J, Johnson MA, Lewis DF (January 1997). "Cytochrome P4501A (CYP1A) induction in rat and man by the benzodioxino derivative, fluparoxan". Xenobiotica. 27 (2): 159–173. doi:10.1080/004982597240668. PMID 9058530.
- Belmaker RH, Agam G (January 2008). "Major depressive disorder". New England Journal of Medicine. 358 (1): 55–68. doi:10.1056/NEJMra073096. PMID 18172175.
External links
 Media related to Fluparoxan at Wikimedia Commons
Media related to Fluparoxan at Wikimedia Commons